
[Genética clásica] [Evolución] [Genética moderna] [Taxonomía] [Geología]
Índice
Índice
Portada

Portada
1. Introducción
1. Introducción
Los genetistas mendelianos entre 1890 y 1910 descubrieron las reglas algebraicas que gobiernan la transmisión de los rasgos genéticos y su relación con los cromosomas y una unidad explicativa fundamental a la cual se le dio el nombre de gen. El gen por su parte fue blanco de muchos tipos de conceptualizaciones, pues no eran pocos los que abogaban por una naturaleza vitalista de los genes, sin embargo, gracias a los trabajos del grupo de investigación de Thomas Hunt Morgan un fenómeno se hizo evidente, fuera lo que fuesen los genes, estos se encontraban almacenados en un lugar físico invariante al interior de los cromosomas, lo que daba la oportunidad para poder crear mapas de los genes aun sin saber de qué estaban hechos. Aun así, el mismo Morgan en 1934 aceptaba que la genética postmendeliana podía operar muy bien sin la necesidad de aclarar si el gen era meramente un artefacto hipotético o una estructura con una materialidad química. Este hecho recuerda en la historia de la química como en su mayor parte, la teoría termodinámica fue construida sin la necesidad de conocer si los átomos eran entidades reales con una estructura interna. Para la década de los 40s las viejas actitudes vitalistas en la biología comenzaron a minarse, y en contra de ciertos filósofos de la ciencia que abogaban por una independencia disciplinar, la comunidad de biólogos y químicos comenzaron a realizar una pregunta incómoda:
¿Cuál es la naturaleza química del gen?
Hacer esta pregunta permitió plantear una serie de experimentos que corroboraron una serie de propuestas y descubrimientos que ya se venían realizando desde el siglo XIX y que llegaron a su punto culminante cuando en 1953 se formula finalmente un modelo para la estructura del ADN, el cual sigue vigente hasta nuestros días, y no es otro que el modelo de la doble hélice. Pero para eso, primero debió demostrarse que los genes estaban hechos de ADN y no de otros materiales químicos.
El centro de los estudios en biología molecular está compuesto por dos grandes tipos de moléculas, por un lado, están las proteínas y por otro los ácidos nucleicos. Aunque actualmente se sabe que los ácidos nucleicos, específicamente el Ácido desoxiriboNucleico o ADN, son los portadores de la información genética, históricamente no fue tan claro. De hecho, el debate sobre la molécula portadora de la información genética se extendería casi durante más de la primera mitad del siglo XX.
En cualquier caso, por el momento solo referiremos una muy leve definición de los ácidos nucleicos ya que son centrales para las discusiones futuras. Los ácidos nucleicos son polímeros, es decir, moléculas muy grandes que se forman como si fueran grandes cadenas. Los dos ácidos nucleicos encontrados en los seres vivos y en los virus son el ADN y el ARN. El ADN es más largo y estable, pero no interactúa con los sistemas moleculares para la creación de proteínas, y por el contrario el ARN “Ácido RiboNucleico” si puede realizar esta interacción, pero por lo general es más corto que el ADN y lo almacena la información genética por leves periodos de tiempo.
Rosalind Elsie Franklin
Rosalind Elsie Franklin (1920-1958) fue una química y cristalógrafa inglesa conocida por su trabajo en la estructura del ADN. Durante su carrera, Franklin desarrolló técnicas innovadoras en cristalografía de rayos X, las cuales le permitieron hacer importantes contribuciones a la comprensión de la estructura de los materiales biológicos. A pesar de su papel fundamental en la investigación del ADN, sus logros fueron minimizados en su tiempo y solo fue reconocida póstumamente.

Contexto social
Rosalind Elsie Franklin nació en 1920 en Londres, Inglaterra, en una época de cambios significativos en el contexto político, económico, social, religioso y cultural del Reino Unido. En el ámbito político, la década de 1920 fue una época de recuperación después de la Primera Guerra Mundial, pero también se caracterizó por la inestabilidad política y la agitación laboral.
En el ámbito económico, el Reino Unido estaba en un periodo de transición a una economía más industrializada y de servicios, lo que resultó en una mayor movilidad social y una creciente clase media.
En el ámbito social, el movimiento sufragista estaba en pleno auge, y las mujeres como Franklin luchaban por sus derechos y reconocimiento en la sociedad. También había una creciente conciencia sobre las desigualdades raciales y de clase, y el país estaba lidiando con la inmigración y la integración de grupos minoritarios.
En el ámbito religioso, la religión anglicana seguía siendo la religión predominante en el Reino Unido, pero también había un aumento en la diversidad religiosa y un declive en la asistencia a la iglesia.
En cuanto a la cultura, la década de 1920 fue conocida como la "era del jazz" y estuvo marcada por una mayor libertad artística y cultural, así como por la creciente influencia de los medios de comunicación y la cultura popular. Todo esto influiría en el desarrollo personal y profesional de Franklin.
Infancia
Rosalind Elsie Franklin nació en Londres, Inglaterra, el 25 de julio de 1920. Fue la segunda hija de una familia judía de clase alta y creció en un ambiente intelectual y educativo privilegiado. Su padre, Ellis Franklin, fue un destacado abogado especializado en derecho fiscal, mientras que su madre, Muriel Waley, era una mujer de origen aristocrático que se dedicaba a la filantropía.
Durante su infancia, Rosalind era conocida por ser una niña muy inteligente y curiosa, con un gran interés por la ciencia y la naturaleza. A los 15 años, ingresó en la Universidad de Cambridge, donde estudió química y se graduó con honores en 1941.
A pesar de que creció en un ambiente privilegiado, la vida de Rosalind no estuvo exenta de dificultades. En su adolescencia, perdió a su mejor amiga debido a una enfermedad, lo que la afectó profundamente. Además, su relación con su madre era a veces tensa, ya que Muriel esperaba que Rosalind se casara y formara una familia, mientras que ella estaba más interesada en su carrera científica. A pesar de esto, Rosalind perseveró y siguió trabajando duro para alcanzar sus objetivos.
Logros
Después de graduarse en química en la Universidad de Cambridge, Rosalind Elsie Franklin trabajó en la investigación química durante la Segunda Guerra Mundial, enfocada en la fabricación de carbón activado para el filtrado de gas. Posteriormente, continuó su formación en física, obteniendo su doctorado en la Universidad de Cambridge en 1945 con una tesis sobre la difracción de rayos X en el estudio de la estructura de carbones y grafitos.
En 1951, Franklin comenzó a trabajar en el King's College de Londres en el departamento de física, donde realizó una serie de investigaciones innovadoras en la técnica de difracción de rayos X, lo que le permitió estudiar la estructura de materiales biológicos como el ADN y las proteínas. Entre sus principales logros científicos destacan:
(a) El descubrimiento de la forma helicoidal de las fibras de ADN, demostrando que el ADN no era una molécula lineal sino que estaba organizada en una estructura en forma de doble hélice.
(b) El desarrollo de técnicas de difracción de rayos X para estudiar la estructura del carbón, virus y proteínas, incluyendo el virus del mosaico del tabaco.
(c) La publicación de varios artículos científicos en revistas importantes, incluyendo una serie de artículos en la revista Nature en los que se describía la estructura del ADN.
Desafortunadamente, los logros de Rosalind Franklin fueron minimizados en su época debido a su papel secundario en la investigación del ADN, en comparación con Watson y Crick, quienes fueron galardonados con el Premio Nobel en 1962 por su descubrimiento de la estructura del ADN. Sin embargo, con el tiempo se ha reconocido la importancia de los logros de Franklin en la comprensión de la estructura del ADN y su contribución a la investigación científica en general.
Comunidad científica
Rosalind Elsie Franklin se desenvolvió en una comunidad científica altamente competitiva, con investigadores que trabajaban arduamente por descubrir los secretos del ADN. Algunos de sus principales colaboradores y oponentes incluyen:
(a) Maurice Wilkins: compañero de trabajo de Franklin en el King's College de Londres, quien también investigaba la estructura del ADN utilizando la técnica de difracción de rayos X. A pesar de que trabajaron juntos, hubo tensiones en su relación profesional y se reportaron algunos desacuerdos en cuanto a la interpretación de los datos.
(b) James Watson y Francis Crick: Investigadores en la Universidad de Cambridge que también estaban interesados en descubrir la estructura del ADN. Si bien inicialmente no colaboraron directamente con Franklin, se cree que se basaron en algunos de sus datos para desarrollar su modelo de la doble hélice del ADN. Esta situación ha llevado a discusiones sobre el reconocimiento adecuado que se le debe otorgar a Franklin por su papel en el descubrimiento de la estructura del ADN.
(c) Linus Pauling: Químico estadounidense que también estuvo interesado en la estructura del ADN, y quien propuso un modelo incorrecto para la estructura de la molécula en 1953.
A pesar de que hubo fricciones y desacuerdos con algunos de sus colegas, Franklin era reconocida por su capacidad de trabajar independientemente y por su habilidad en la investigación científica. A pesar de que su trabajo fue a menudo minimizado en su época, con el tiempo se ha reconocido su importancia en el descubrimiento de la estructura del ADN y en la investigación científica en general.
Reconocimientos
A pesar de que Rosalind Elsie Franklin no recibió muchos reconocimientos en vida, su trabajo ha sido reconocido y honrado póstumamente en varias formas. Algunos de los reconocimientos más importantes incluyen:
(a) En 1956, fue elegida miembro de la Royal Society, una de las más prestigiosas organizaciones científicas del mundo.
(b) En 1962, Watson, Crick y Wilkins fueron galardonados con el Premio Nobel de Fisiología o Medicina por su descubrimiento de la estructura del ADN. Aunque Franklin no recibió el Premio Nobel, muchos en la comunidad científica han expresado su descontento por la omisión de su trabajo en la concesión del premio.
(c) En 1995, la Royal Society creó la Medalla Rosalind Franklin, en su honor. Esta medalla se otorga cada dos años a una mujer científica que haya realizado una contribución significativa en cualquier campo de las ciencias naturales.
(d) En 2004, el King's College de Londres colocó una placa en su memoria en su lugar de trabajo en la universidad.
(e) En 2013, la Universidad de Cambridge nombró a una de sus residencias estudiantiles en honor a Franklin.
(f) En 2018, el Consejo Europeo de Investigación (ERC) otorgó una beca de investigación de 2.5 millones de euros para un proyecto en el que se utiliza la técnica de difracción de rayos X para estudiar la estructura de proteínas, en honor al trabajo de Rosalind Franklin.
Estos honores y reconocimientos reflejan el impacto duradero de la investigación de Rosalind Franklin en la comprensión de la estructura del ADN y su legado en la investigación científica.
¿Por qué ella es importante?
El trabajo de Rosalind Franklin en la estructura del ADN ha tenido un impacto significativo en nuestra vida cotidiana en muchos aspectos.
En primer lugar, el ADN es la base de la biología moderna y la comprensión de su estructura ha llevado a importantes avances en la medicina, la biotecnología y la biología molecular. Gracias a la comprensión de la estructura del ADN, se han desarrollado técnicas para identificar y tratar enfermedades genéticas, se han creado nuevos tratamientos y terapias, y se han descubierto muchas de las bases moleculares de la vida.
Además, la investigación de Franklin en la técnica de difracción de rayos X ha sido fundamental para el estudio de estructuras moleculares en general. Hoy en día, esta técnica se utiliza en muchos campos diferentes, desde la química hasta la biología y la física, para determinar la estructura de moléculas importantes como las proteínas, los lípidos y los carbohidratos.
En resumen, el trabajo de Rosalind Franklin ha sentado las bases para muchos de los avances en biología y medicina de las últimas décadas, y ha permitido el desarrollo de nuevas tecnologías y terapias que mejoran nuestra calidad de vida y prolongan nuestra esperanza de vida.
¿Qué debemos aprender de ella?
La vida de Rosalind Franklin es un ejemplo de perseverancia, determinación y dedicación a la ciencia. A pesar de las dificultades y barreras que enfrentó en su carrera científica, nunca perdió su pasión por la investigación y logró importantes avances en su campo.
Uno de los mayores obstáculos que enfrentó fue el sesgo de género en el mundo académico y científico de la época. A pesar de esto, continuó trabajando duro y haciendo importantes contribuciones a su campo, a menudo sin recibir el reconocimiento que merecía.
A pesar de las limitaciones que enfrentó, Rosalind Franklin mantuvo su enfoque en su trabajo y logró grandes avances en la comprensión de la estructura del ADN. Su impacto en la biología y la medicina ha sido duradero y ha ayudado a sentar las bases para muchos de los avances que disfrutamos hoy en día.
La vida de Rosalind Franklin nos muestra la importancia de no rendirse ante los obstáculos y de seguir nuestra pasión, incluso en situaciones difíciles. Además, su ejemplo nos enseña la importancia de no subestimar el impacto que podemos tener en el mundo, incluso si no recibimos el reconocimiento que merecemos. Al perseverar y seguir trabajando duro, podemos hacer contribuciones significativas y tener un impacto duradero en nuestro campo y en el mundo en general.
2. Naturaleza química del gen
2. Naturaleza química del gen
Con esto la afirmación es simple, los genes son sustancias químicas.
Aislamiento del ADN
Como materiales, los químicos ya habían logrado el aislamiento de ambos ácidos nucleicos “mezclados ambos” de la célula y el núcleo. La naturaleza polimérica era fácil de apreciar a un nivel cualitativo, de hecho, el ADN tiende a formar fibras transparentes-blanquecinas cuando es aislado. Friedrich Miescher “1844–1895” fue el primero en aislar la mezcla de ácidos nucleicos en 1869 dándole el nombre de nucleina. Tiempo después el aisló una muestra pura del material conocido actualmente como ADN en los espermatozoides de salmón, y en 1889 Richard Altmann “1852-1900” le dio el nombre de ácido nucleico por su capacidad para ceder protones, esta capacidad se debe a la presencia de grupos fosfato, los cuales en libertan forma en ácido fosfórico. También quedó claro que este ácido nucleico solo se encontraba en los cromosomas.

Figura 2.1. ADN aislado.
En 1919 Phoebus Levene en el instituto Rockefeller identificó los componentes químicos de dicho material, se trataba de un polímero complejo compuesto por cuatro bases nitrogenadas, dos de ellas clasificadas como purinas y las otras dos como pirimidinas. Adicionalmente se encontró un azúcar de cinco carbonos y grupos fosfato, los cuales formaban las uniones del polímero.

Figura 2.2. De izquierda a derecha: Friedrich Miescher, Phoebus Levene, y Nicolai Koltsov.
En 1919 Phoebus Levene en el instituto Rockefeller identificó los componentes químicos de dicho material, se trataba de un polímero complejo compuesto por cuatro bases nitrogenadas, dos de ellas clasificadas como purinas y las otras dos como pirimidinas. Adicionalmente se encontró un azúcar de cinco carbonos y grupos fosfato, los cuales formaban las uniones del polímero.
Levene llamó a cada una de estas unidades “nucleótido” y sugirió qué la molécula del ADN consistía en una cadena de nucleótidos unidos a través de los grupos fosfato, los cuales formarían el esqueleto sobre el cual se anclarían las bases nitrogenadas. Sin embargo, Levene pensaba que las cadenas no podrían ser muy largas, y que las bases se presentarían repetidas de formas fijas. Otros autores que demostraron la naturaleza polimérica del ADN anticipada por Levene fueron Torbjorn Caspersson y Einar Hammersten.
En 1927 Nicolai Koltsov "1872-1940" propuso que los rasgos heredables serían heredades de una generación a la siguiente por medio de una molécula de almacenamiento de la información genética gigantesca, la cual debía estar formada por dos unidades idénticas como si fueran imágenes en un espejo. Estas dos moléculas podrían entonces replicar su otra cara en la fase de síntesis del material genético durante el ciclo celular, de forma tal que la mitad del material genético se conservara y la otra mitad fuera el nuevo. En otras palabras, la replicación semi-conservativa fue propuesta como una de las características que debía poseer esta hipotética molécula de almacenamiento de la información. Posteriormente Max Delbrück "1906-1981", Nikolai V. Timofeeff-Ressovsky "1900-1981", y Karl G. Zimmer "1911-1988" publicaron en 1935 sugirieron que los cromosomas no eran más que, enormes moléculas que podían experimentar reacciones químicas mediante el bombardeo con radiación de ultra-alta energía como los rayos X, y por lo tanto, su estructura podía cambiar. Como una consecuencia de lo anterior, proponían que los rasgos heredables podían ser alterados por medio del bombardeo con rayos X, o lo que es lo mismo, los rayos X servían para inducir mutaciones. En 1937 William Astbury "1898-1961" realizó un avance fundamental, perfeccionando el método que permitiría acumular los datos experimentales necesarios para la elucidación de la estructura del ADN, y no es otra cosa que la difracción con rayos X de la molécula del ADN, lo cual al ser revelados ante una placa generaba una serie de patrones. El problema es que no fue capaz de deducir un modelo de estructura del ADN que explicara los patrones de la placa de revelado.
Imtroducción al experimento Avery–MacLeod–McCarty
El experimento Avery–MacLeod–McCarty es uno de los experimentos más famosos en la biología molecular, y se reporta como la primera demostración de que el ADN tiene algo que ver con los rasgos heredables al interior de una célula. Fue reportado en 1944 por Oswald Avery "1877-1955", Colin MacLeod "1909-1972", y Maclyn McCarty "1911-2005", empleando el fenómeno de la transformación bacteriana. Durante la época ya se conocían los ácidos nucleicos y las proteínas, así como su estructura bioquímica. La mayoría de la comunidad científica daba más crédito a la hipótesis de que las proteínas eran el material genético debido a que eran más complejas que los ácidos nucleicos. Este experimento fue la culminación de varias investigaciones realizadas desde los años de 1930 en el Instituto de Investigación Médica Rockefeller con el fin de purificar cierto factor que inducia a la transformación de cepas de bacterias inocuas, en variantes mortales capaces de causar neumonía fatal.

Figura 2.3. Fila superior: William Astbury y Oswald Avery; fila inferior: Colin MacLeod y Maclyn McCarty.
Como se ha mencionado anteriormente, en la época desde inicio del siglo XX y durante casi los siguientes 50 años la mayor parte de la comunidad científica pensaba que las proteínas y no los ácidos nucleicos, eran las responsables del almacenamiento de la información genética, es decir, las proteínas eran el fundamento químico del gen de los mendelianos y postmendelianos. Esto se sostenía bajo el argumento de que los ácidos nucleicos eran demasiado simples como para almacenar la información genética necesaria para la construcción de las proteínas, por lo que no es descabellado asumir que la función de organelos, como el ribosoma, aún era desconocidas.
Un experimento previo realizado por Griffith en 1928 planteaba el asunto. El mató una cepa de bacterias virulentas de Streptococcus pneumoniae de la cepa III-S que era capaz de causar neumonía fatal en los ratones. Luego, mezclo los remanentes de estas bacterias cuyas células ya se habían destruido en un ambiente con una cepa no virulenta de S. pneumoniae del código II-R. El resultado fue que las cepeas no virulentas se transformaron en la cepa virulenta por medio de algún mecanismo en los restos muertos de la primera cepa virulenta.

Figura 2.4. Experimento de Griffith. Resumen del experimento de Griffith. de izquierda a derecha (1) la cepa no virulenta no es mortal, (2) la cepa virulenta es mortal, (3) la cepa virulenta tratada con calor no es mortal, (4) la cepa no virulenta con los restos muertos de la cepa virulenta es mortal, y al extraerla posee un serotipo y propiedades de cepa virulenta.
¿Qué es lo que causa la transformación? El experimento Avery–MacLeod–McCarty en su mismo título plantea la hipótesis de trabajo: Estudios sobre la naturaleza química de la sustancia que induce a la transformación de tipos de Pneumococos: Inducción a la transformación por medio de una fracción de ácido desoxirribonucleico extraído de Psenumococo tipo III; publicado en 1944 se convertiría en la primera sugerencia experimental de que el ADN y no las proteínas, eran el verdadero agente que se encargaba del almacenamiento de la información genética.
Marco de referencia del experimento Avery–MacLeod–McCarty

Figura 2.5. Dos cepas de Streptococcus pneumonie. A la izquierda las colonias virulentas lisas con respecto al agar y a la derecha las cepas no virulentas rodeadas por un halo de alfa-hemólisis, agar Columbia con 5% de sangre de cordero defibrinada y al 5% de dióxido de carbono
El experimento Avery–MacLeod–McCarty requirió una serie de desarrollos metodológicos, los cuales incluyen (1) la serotipificación; (2) la demostración de que las bacterias se pueden transformar; (3) los métodos para transformar bacterias en tubos de ensayo; y el más importante (5) los métodos para poder aislar el agente transformador. La serotipificación es un método en el cual se emplean marcados biológicos, en este caso anticuerpos del sistema inmune, para identificar con gran precisión y exactitud diferentes tipos de bacteria de una misma morfoespecie. Estos tipos difieren en muchas de sus propiedades bioquímicas y son denominados cepas, a las cuales se les asigna un código. El científico que lideró estos primeros esfuerzos de tipificación de las cepas por códigos y propiedades fue Fred Neufeld "1869-1945". Originalmente, la tipificación bacteriana cayó bajo un esquema filosófico muy antiguo, el esencialismo o pensamiento tipológico, es decir, ausencia de cambios. Sin embargo, siendo las bacterias los seres vivos más adaptables, no pasó mucho tiempo hasta que otros autores se percataran de que los serotipos bacterianos eran capaces de cambios rápidos y abruptos en sus propiedades biológicas y bioquímicas.

Figura 2.6. La neumonía. La neumonía es una inflamación de los alvéolos y capilares pulmonares, que impide una adecuada ventilación del sistema circulatorio causada por la infección con carios patógenos. Gracias al desarrollo de los antibióticos se pensó que esta y otras enfermedades bacterianas habían sido erradicadas.
El primer bacteriólogo que se dio cuenta de la capacidad de los serotipos para transformarse fue Frederick Griffith "1879-1941", perteneciente a la siguiente generación de científicos después del establecimiento de la metodología de clasificación serológica. El experimento de Griffith “1928” fue realizado no en un cultivo en un tubo de ensayo o en una caja de Petri; por el contrario, se realizó al interior de un modelo biológico, el ratón de laboratorio.
Griffith había pasado varios años estudiando y clasificando diferentes cepas de neumococos, causantes de una enfermedad fatal para la década de los 20s, la neumonía. El primer resultado de sus estudios fue que no todos los neumococos eran iguales, algunos no eran fatales o no causaban ninguna patología, mientras que otras eran fatales. Un resultado inesperado de sus investigaciones en la época fue el hecho de que no todas las cepas se encuentran en un mismo individuo al mismo tiempo, las cepas tendían a excluirse y por lo tanto a generar cuadros de síntomas diferentes.
Otro descubrimiento de le dio la clave de todo, si bien las cepas se excluían, a lo largo de un solo caso clínico, se registraba un cambio en el tipo de cepa aislada, lo cual lo indujo a pensar que (1) una cepa reemplazaba a la otra excluyéndola competitivamente u (2) una cepa se transformaba en otra mediante algún tipo de mecanismo desconocido. Para refutar la hipótesis (1) lo que hizo fue extraer una cepa virulenta de un individuo y matar las bacterias, de esta forma se aseguraba de que no existiera posibilidad para una exclusión competitiva. Luego inyecto los restos muertos de la cepa mortal en un ratón que ya estaba infectado con una cepa no virulenta. El resultado fue la muerte del ratón. Con esto quedaba demostrado que los restos de las bacterias muertas poseían algún factor químico capaz de transformar una cepa no virulenta en una virulenta, lo cual a su vez demostraba que las cepas podían cambiar rápidamente. En otras palabras, las cepas no se excluyen mutuamente, simplemente cuando ocurre una segunda infección con una cepa virulenta, las bacterias mortales transforman a las residentes no virulentas en su forma mortal “a la apocalipsis zombi”.
Los descubrimientos de Griffith no aclararon quien era el agente causante de la transformación, y de hecho cualquier hipótesis era válida, dado que los restos empleados para transformar a la bacteria no virulenta fueron todos los componentes de las bacterias virulentas, lípidos, proteínas, ácidos nucleicos, cualquier cosa. Los resultados de Griffith fueron rápidamente confirmados por Fred Neufeld "1869-1945" del Instituto Koch por Martin Henry Dawson "1896-1945" del Instituto Rockefeller. El grupo de investigación del Instituto Rockefeller continuaron estudiando la transformación de las bacterias. Junto a Richard H.P. Sia, Dawson desarrolló un nuevo método para transformar bacterias en tubos de ensayo. Este descubrimiento abría las puertas para poder controlar con mayor precisión los agentes transformadores, aunque la pregunta seguía abierta, ¿Quién era el agente que transformaba a las bacterias?
Después de que Dawson se marchara en 1930, James Alloway prosiguió en la línea de investigación sobre los serotipos de neumococcos y su capacidad para transformarse de una cepa a otra. Su empeño de vio recompensado cuando en 1933 pudo extraer en solución acuosa una sustancia que prometía ser el agente transformador. Colin MacLeod prosiguió en la misma línea de investigación, buscando purificar todas las sustancias que se encontraban en las soluciones acuosas de Alloway entre los años de 1934 y 1937. Posteriormente al inicio de la década de los 40s su trabajo sería reasumido por Maclyn McCarty. Como se puede evidenciar, el experimento Avery–MacLeod–McCarty no salió de la nada, como todo en ciencia se trata de la genealogía de una idea que es pasada de una generación a otra, de maestro a alumno, de forma tal que el conocimiento de la generación anterior se convierte en los fundamentos de la siguiente, con lo que, parafraseando a Newton y Einstein, en las ciencias, los científicos deben pararse sobre los hombros de los gigantes que les han precedido.
Procedimiento del experimento Avery–MacLeod–McCarty
Personalmente, la primera vez que entré en contacto con el experimento de Avery–MacLeod–McCarty, el experimento se me describió en términos de la inyección de los ratones del agente transformador. Esto da la impresión de que el asunto de la muerte de los ratones por la transformación de la bacteria en su forma virulenta era algo nuevo.

Figura 2.7. Procedimiento del experimento Avery–MacLeod–McCarty. Este se basa en extraer los componentes celulares y luego eliminarlos uno a uno hasta que el ratón vivo.
Sin embargo, como hemos visto anteriormente, el asunto de la transformación de las cepas de neumococcos era una línea de investigación con una larga tradición al interior de algunas comunidades de investigación, por lo tanto, cabe preguntarse, ¿Cuál es el aporte real del experimento Avery–MacLeod–McCarty? El punto fue la purificación, aislamiento y caracterización del agente transformador en solución acuosa, y la demostración que, mediante la administración de este agente aislado y puro en una cepa no virulenta, esta se convertía en su forma virulenta. Por tal razón, el procedimiento experimental en su mayor parte recuerda más a la química, en el sentido de que se trata de purificar una molécula e identificarla por medio de la comparación con una muestra patrón.
El procedimiento de purificación que Avery realizó consistió en matar las bacterias mediante intenso calor y extraer los componentes mediante una solución salina. Esto genera una mezcla de componentes solubles como carbohidratos, proteínas hidrosolubles y ácidos nucleicos, pero excluye a las proteínas liposolubles y a los lípidos. Posteriormente, las proteínas hidrosolubles fueron precipitadas con cloroformo, y los carbohidratos complejos de las capsulas se hidrolizaron por medio de una enzima. Posteriormente, para evitar reacciones cruzadas con alguna proteína de la capsula se emplearon enzimas específicas para completar su destrucción. Finalmente, la sustancia activa fue separada de la solución acuosa por medio de fraccionamiento con alcohol que la hace insoluble, lo cual genera fibras blancas-transparentes que pueden ser removidas con facilidad por medios físicos “igual que hilar un hilo”. La identificación de la sustancia reveló consistencia con las muestras patrón del ácido desoxiribonucleico. Esto mostraba que era ADN y no otras sustancias como ARN o proteínas las que representaban la composición mayoritaria de la muestra aislada. A pesar de esto, la muestra purificada fue tratada del siguiente modo. Una muestra no fue tratada con ninguna enzima, siendo así la muestra control. Otra muestra fue tratada con proteasas como la tripsina y la quimotripsina, con el objeto de descomponer cualquier resto de proteínas. Otra muestra fue tratada con ribonucleasa, una enzima que corta al ácido ribonucleico de forma específica. La última muestra fue tratada con desoxiribonucleasa, una enzima que corta de forma específica al ácido desoxiribonucleico. Estas muestras fueron mezcladas con un cultivo de bacterias de pnemucocos no virulentos, y posteriormente se determinaron las propiedades bioquímicas de los cultivos en agares especializados. La única de las muestras que no mostró haber transformado a las bacterias fue la tratada con la desoxiribonucleasa, mientras que las demás mantuvieron su actividad.
Legado del experimento Avery–MacLeod–McCarty
Los hallazgos experimentales del experimento de Avery–MacLeod–McCart fueron rápidamente confirmados y amplificados por otras características hereditarias además de la virulencia. Aun así, la mayor parte de la comunidad científica aún mantenía sus dudas sobre la metodología empleada, específicamente en lo que respecta a la purificación de la muestra de todas las proteínas. Esto se debió en gran parte a que la propuesta para la estructura del ADN vigente en la época se basaba en la hipótesis de los tetranucleótidos, es decir, repeticiones de a cuatro bases nitrogenadas, lo que convertía al ADN en una molécula incapaz de almacenar la información biológica. Una línea de evidencia que reforzaba la hipótesis de que las proteínas eran el material hereditario eran los virus, quienes habían sido determinados químicamente como compuestos por proteínas por Wendell Stanley “1904-1971”. Algunos biólogos dudaban que la genética desarrollada en base a las leyes de Mendel y sus extensiones pudiera ser aplicada a las bacterias debido a que estas carecían de los cromosomas o de una reproducción sexual. A pesar de que este experimento representa una de las primeras aproximaciones formales a la función del ADN su impacto fue muy limitado, y su celebración como parte de la historia de la bacteriología solo se dio 9 años más tarde cuando la función del ADN como material genético fuese confirmada por el experimento de Hershey-Chase.
3. Las reglas de Chargaff
3. Las reglas de Chargaff
Para comprender como funciona una molécula muy compleja, sea esta una proteína, un lípido, un carbohidrato o un ácido nucleico, es necesario en primera instancia conocer sus partes constitutivas.
El reduccionismo en biología
Este modo de estudiar la biología es denominado reduccionismo y es atacada por lo que queda de filósofos de la ciencia que se autodenominan vitalistas. Reducir la vida a un montón de moléculas resulta incoherente con las capacidades únicas de los seres vivos, cuando se las compara con estructuras inorgánicas. Esta tendencia llevó a que en tiempos de Arhenius y Wohler se dudara de la posibilidad de sintetizar moléculas de importancia orgánica a partir de precursores inorgánicos. Sin embargo, el método reduccionista no es malo en sí mismo desde que no se asuma que el todo tiene las mismas propiedades que sus partes sumadas. Estudiar las partes es el punto de partida, y de cierta forma permite adivinar como algunas de estas partes se relacionan para generar propiedades emergentes de las estructuras como un todo. Esto es lo que diferencia la postura organicista, que el humilde escritor de este texto asume como postura epistemológica en biología. Una postura que busca congeniar el reduccionismo y el holismo en un solo marco explicativo a través del concepto de propiedad emergente.
Propiedades emergentes
Las propiedades emergentes son características o comportamientos que surgen en un sistema complejo como resultado de la interacción y organización de sus componentes individuales. En otras palabras, las propiedades emergentes son propiedades que no se pueden explicar simplemente observando los componentes individuales de un sistema (un sinónimo de esto es no-superveniencia), sino que se derivan de la forma en que interactúan.
El ADN es un excelente ejemplo de un sistema que exhibe propiedades emergentes. El ADN está compuesto de nucleótidos individuales, cada uno de los cuales tiene una estructura y función específicas. Sin embargo, la interacción y organización de estos nucleótidos en una doble hélice permite que el ADN exhiba propiedades emergentes, como la capacidad de almacenar y transmitir información genética.
Otra propiedad emergente del ADN es su capacidad para plegarse y adoptar diferentes formas tridimensionales, lo que permite que interactúe con proteínas y otros componentes celulares. Esta capacidad de plegamiento es una propiedad emergente que surge de la interacción entre los nucleótidos individuales que componen el ADN.
Las propiedades estructurales del ADN son emergentes con respecto a sus componentes, es decir, cada componente o sección del ADN carece de las propiedades de la molécula completa, pero, aun así, su estudio es relevante para comprender la naturaleza de dichas propiedades emergentes totales.

Figura 3.1. Componentes de un nucleótido. Izquierda: modelo molecular de la desoxiribosa, se denomina así porque en el carbono 2 no se encuentra un grupo hidroxilo, sino un simple protón, en base a esta falta de un oxígeno se la llama ribosa desoxígenada o desoxirribosa; Centro: La ribosa y un fosfato, por lo general los fosfatos se unen en el carbono 3, pero también pueden unirse por el carbono 5; y Derecha: el núcleótido adenocina o AMP.
Las bases nitrogenadas
Las bases nitrogenadas que Levene y sus sucesores encontraban respondían a dos grupos diferentes, llamados purinas y pirimidinas. La base nitrogenada se unía al azúcar por el extremo 3´. Las pirimidinas contienen un único anillo de carbonos, mientras que las purinas contienen dos anillos de carbonos.

Figura 3.2. Bases nitrogenadas. Las purinas son de doble anillo, mientras que las pirimidinas son de anillo simple.
El ADN contiene dos tipos diferentes de pirimidinas, la timina y la citosina, ambas simbolizadas por su primera letra (T) y (C) respectivamente. Las purinas también son de dos tipos, la guanina (G) y la adenina (A), los nucleótidos se unen de forma covalente mediante los residuos de fosfato que se unen entre los carbonos 5´y 3´formando un polímero linear o hebra, a este enlace se lo denomina fosfodiester. No hay que confundir las bases nitrogenadas con los nucleótidos, un nucleótido es una estructura compuesta por azúcar+base nitrogenada, y como tal poseen nombres moleculares que los distinguen.
Las bases nitrogenadas se unen a cada azúcar, cuando el azúcar se polimeriza a través del enlace fosfodiester, las bases nitrogenadas se proyectan como los escalones de una escalera, mientras que el esqueleto de la escalera permanece de forma externa al cilindro, todos los átomos de oxígeno le confieren a la molécula una alta solubilidad por poder entablar puentes de hidrógeno. Las posiciones para la polimerización del azúcar son en el carbón 3´y 5´, detalle que se convertirá en fundamental cuando se propongan los modelos de replicación del ADN. Debido a que cada hebra es complementaria y antiparalela, significa que una termina en el extreme 3´y termina en el 5, mientras que su complemente actúa de forma inversa iniciando en el extremo 5´y terminando en el 3´. Los datos de la difracción con rayos X indicaba que la distancia entre los nucleótidos era de 3.4 Armstrong o 0.34 nanómetros, y sugerían la presencia de una estructura repetitiva cada 3.4 nanómetros.
Organización de las bases nitrogenadas
Levene pensaba que las bases nitrogenadas se organizaban como un tetrámero repetitivo “en tándem” de la forma 3´-ATGC ATGC ATGC ATGC ATGC ATGC-5´. Esta falta de variabilidad ubiera hecho imposible que el ADN fuera la naturaleza química del gen. En 1950, dos años antes del experimento Hershey-Chase otra línea de evidencia en favor del ADN fue abierta por Erwin Chargaff “1905-2002” de la universidad de Columbia reportó los resultados de un importante experimento, el cual le daría el golpe final a la teoría de Levene de los tetranucleótidos.

Figura 3.3. Erwin Chargaff (11 de agosto de 1905 – 20 de junio de 2002) Fue un químico austriaco judío que emigró a los Estados Unidos durante la anexión nazi de su país. Después de cuidadosos experimentos, Chargaff descubrió dos reglas que ayudaron al descubrimiento de la doble hélice del ADN.
Chargaff determinó la cantidad relativa de bases nitrogenadas en diferentes muestras de ADN. El análisis de composición fue realizado mediante la acción de una enzima que corta la unión entre las bases nitrogenadas y los azucares, posteriormente, la muestra fue separada mediante una cromatografía en papel generándose cuatro puntos de migración. Las cantidades de sustancia podían interpretarse de forma relativa de acuerdo al tamaño del punto de migración “básicamente el tamaño de la mancha”.
Si la hipótesis de los tetranucleótidos de Levene fuera correcta se esperaría cuatro manchas, cada una con una forma semejante debido a su cantidad semejante “cada una debía representar un 25% de la muestra total”. Los resultados de Chargaff fueron muy diferentes de lo que se esperaba en base a la hipótesis de Levene, es más, cada muestra de ADN mostraba su propia composición única, la cual difería radicalmente de los radios 1-1-1-1. Sin embargo, había que aclarar el tipo de muestra.
Las reglas
Cuando la muestra provenía de diferentes especies, las proporciones eran variables, pero cuando la muestra provenía de la misma especie, las proporciones eran semejantes.
Tabla 3.1. Composición porcentual (%) del ADN de varias especies.

A pesar del aspecto variable de la composición del ADN en términos de sus bases nitrogenadas, emergió un fenómeno que era constante.
El número de adeninas siempre era igual al número de timinas; mientras que el número de guaninas siempre era igual al número de citosinas.
Los descubrimientos de Chargaff arrojaron nuevas luces sobre la estructura de la molécula del ADN, ya no sería vista como una molécula invariante, aunque al igual que con otros descubrimientos relacionados al ADN antes de 1952 permaneció oscuro en la literatura científica hasta el gran bum del ADN entre los años de 1952-1953. Todo lo anterior implica que cada una de las líneas de evidencia sobre la importancia del ADN por si solas no aportaban un mayor peso a la teoría general, pero una vez que tuvo todo el panorama, estas investigaciones que habían permanecidos oscuras por muchos años, de un momento a otro fueron celebradas por la comunidad científica. En otras palabras, una vez que un programa de investigación se demuestra victorioso, todos sus descubrimientos de un momento a otro se hacen importantes, aun cuando en sus verdaderos contextos no se hubieran tomado realmente en cuenta.
4. El experimento Hershey–Chase
4. El experimento Hershey–Chase
En realidad, se trata de una serie de experimentos realizados en 1950 por Alfred Hershey “1908-1997” y Martha Chase “1927-2003” que ayudaron a confirmar que el ADN es el material que almacena la información hereditaria que se transfiere de una generación a otra. Como hemos visto en series anteriores, los químicos y biólogos habían extraído, purificado y determinado químicamente el ADN ya desde finales del siglo 1869, pero debido a la complejidad estructural de las proteínas, la mayor parte de la comunidad científica aun asumía en 1952 que la molécula que almacenaba la herencia era otra proteína.

Figura 4.1. Martha Cowles Chase (1927 – 8 de agosto de 2003). También conocida como Martha C. Epstein, fue una bióloga estadounidense especializada en genética, famosa mundialmente por haber formado parte del grupo que en 1952 demostró que el ADN es el material genético para la vida, y no las proteínas.
Aunque también se conocía que el ADN hacía parte de los cromosomas, se lo consideraba más como un componente estructural y estabilizador que otra cosa. Aunque los autores fueron cautelosos en sus conclusiones, otros experimentos posteriores confirmaron sus resultados, y por consiguiente la función del ADN como la molécula encargada del almacenamiento de la información genética fue elevada a los mayores niveles de certidumbre en las ciencias de la naturaleza. Esto sucedió tan solo un año antes de que se propusiera el modelo para la estructura molecular del ADN.

Figura 4.2. Alfred Day Hershey (4 de diciembre de 1908 - 22 de mayo de 1997). Químico y se doctor en Bacteriología en 1934 en la Universidad de Míchigan. En 1950 se traslada al Instituto Carnegie en el departamento de genética en Washington. En 1952, junto a Martha Chase, confirman que es el ADN la base del material genético, y no las proteínas. Este trabajo será recordado como el experimento de Hershey y Chase.
Marco de referencia
Desde épocas antiguas ha sido registrada la capacidad de las aguas de ciertos ríos para curar enfermedades de forma casi milagrosa como la lepra. En 1896 Ernest Hanbury Hankin reportó que algo en las aguas de los ríos Ganges y Yamuna en la india poseían una marcada propiedad antibacteriana contra el cólera, esta propiedad podía traspasar incluso los filtros de porcelana. En 1915 el bacteriólogo británico Frederick Twort “1877-1950” superintendente del Instituto Brown de Londres, descubrió un pequeño agente que infectaba y mataba las bacterias, lamentablemente la I guerra mundial impidió que sus investigaciones prosiguieran su curso. De forma independiente el microbiólogo Franco-Canadiense Félix d'Hérelle, quien trabajaba en el Instituto Pasteur de París anunció el 3 de septiembre de 1917 que había descubierto “un microbio invisible que era antagonista a los patógenos que causaban la disentería”. La conclusión a la que llegó d´Hérelle fue que se trataba de un virus que mataba las bacterias. El nombre que se le dio fue el de bacteriófago “devorador de bacterias” (YouTube). Inicialmente se pensaba que los virus eran compuestos únicamente de proteínas, por lo que la investigación realizada por ciertos miembros de la comunidad científica conocidos informalmente como el grupo de los fagos conllevaría a confirmar que el ADN es el material genético, no solo al interior de los fagos, sino en términos más generales de todos los seres vivos.
Procedimiento experimental
Hershey y Chase necesitaban ser capaces de examinar diferentes partes de los fagos de forma separada, por lo que era importante separar las secciones estructurales del virus. Para la época ya se sabía que los virus poseían ADN y una serie de proteínas a su alrededor. ¿La solución? Evidentemente los científicos no tienen cámaras moleculares para poder “ver” con los ojos lo que sucede al nivel molecular, es allí donde la imaginación, el ingenio y la lógica entran a trabajar. Recordemos que a pesar de que los virus junto con los seres vivos son entidades totales que exhiben propiedades que van más allá de la suma de sus partes, cada una de sus partes está compuesta por átomos de elementos como el carbono, el nitrógeno, el azufre, el fósforo o el hidrógeno. Los elementos no son entidades homogéneas, cada elemento puede presentarse en diferentes versiones llamadas isótopos, y algunos isótopos emiten radiaciones que pueden detectarse fácilmente con el equipo adecuado. Es decir, es posible marcar cada sección del fago, incluyendo su ADN con diferentes isótopos.
Para distinguir entre proteínas y ADN se emplearon el fósforo y el azufre, esto se debe a que las proteínas no contienen fósforo, mientras que el ADN sí; mientras que todas las proteínas contienen azufre. El nitrógeno no era útil ya que ambas sustancias contienen nitrógeno. Los isótopos elegidos fueron el fósforo-32 para marcar al ADN y el azufre-35 para marcar las proteínas. Incorporar isótopos no se puede realizar de forma manual, para lograrlo se cultivaron bacterias en medios ricos con los isótopos durante unas cuatro horas, de forma tal que sus proteínas y material genético se cargaban con los marcadores. Después de 4 horas, se infectaron estas bacterias con los fagos, de forma tal que estos adquirieron los isótopos marcados en sus propias estructuras. Este procedimiento fue hecho una vez para los fagos marcados con azufre y una vez para los fagos marcados con fósforo. Posteriormente, la progenie marcada fue extraída del caldo de cultivo y transferida a otro medio con isótopos normales, con bacterias sin marcar. Cuando un fago ingresa a una bacteria no lo hace de forma completa, una enorme sección se queda por fuera, la cuestión era saber que material era el que ingresaba a la bacteria, pues evidentemente se trataba del material genético del virus.

Figura 4.3. Procedimiento del experimento Hershey-Chase. El punto de las marcas era ver que material ingresaba a la célula y, por ende, causaba la infección. Durante el experimento el resultado más importante fue que el ADN ingresaba y las proteínas se quedaban a fuera de las células infectadas.
Una vez se generaba la infección, las proteínas que permanecían fuera de la bacteria podían ser removidas por medio de una centrifugación, lo cual permitía tener acceso únicamente al material que había ingresado a las bacterias. Los resultados fueron que, para los casos en que se marcaron las proteínas de los fagos, la segunda progenie emergía sin marcar. Mientras que para el caso en que se marcó el ADN la segunda progenie mantenía el marcador de fósforo radiactivo. La consecuencia era clara, el material que había sido transmitido desde la generación marcada hasta la segunda generación había sido aquella marcada con el fósforo, y dado que no existen proteínas con fósforo, la conclusión forzaba a admitir que
era el ADN el que había sido transmitido entre las dos generaciones.
5. El conflicto por la estructura del ADN
5. El conflicto por la estructura del ADN
Debido a su enorme importancia social, la gente espera que los científicos sean algo así como personas con valores éticos por encima del promedio casi cercano a La Santidad. Sin embargo, la realidad es diferente, los científicos somos seres humanos, y susceptibles a todas sus pasiones y bajezas.
La historia de muchos descubrimientos científicos es también la historia y el drama y conflictos de prioridades de muchos científicos por obtener la gloria y la fama eternos, y la historia del descubrimiento de la estructura del ADN es uno de los capítulos más dramáticos.
Linus Pauling
Las décadas entre los años 30s y mitad de los 50s del siglo XX fueron un caldo de cultivo para una nieva comunidad científica basada en el estudio de la biología a un nivel molecular basada en los instrumentos y la lógica de la química e inclusive de la física. Tal vez no existe autor que pueda conglomerar de forma tan especializada los tres grandes campos de las ciencias de la naturaleza como Linus Pauling “1901-1994”, el mayor genio que hubieran dado los químicos en el siglo XX. Pauling generalmente es mencionado debido a su principio de exclusión en la mecánica cuántica, pero lo que pocos saben es que su laboriosa mente tocó de forma profunda a la biología molecular, no solo en su empeño por lograr descubrir la estructura de las proteínas, sino también la del ADN.

Figura 5.1. Primera fila: Linus Pauling y Robert Corey; segunda fila: Herman Branson y Sir Lawrence Bragg.
A mediados de los años 30s, Pauling, fue influenciado fuertemente hacia la biología debido a las prioridades económicas de la fundación Rockefeller, por lo que decidió investigar en las nuevas áreas de moda. A pesar de que ya había realizado un trabajo monumental en la estructura de las moléculas inorgánicas, y especialmente en la naturaleza del enlace químico visto a la luz de la teoría mecánico-cuántica, él había explorado de forma esporádica algunas moléculas de interés biológico en parte debido al reciente auge de la biología en el Instituto Técnico de California “Caltec”. Pauling interactuó con grandes biólogos como T. H. Morgan “1866-1945”, T. Dobzhansky “1900-1975”, C. Bridges “1889-1938” y A. Sturtevant “1891-1970”. Sus primeros trabajos en el área biológica se relacionaron con la estructura de la hemoglobina. Pauling demostró que la molécula de hemoglobina cambia su estructura cuando gana o pierde un átomo de oxígeno. Como resultado de esta observación, el decidió conducir más investigaciones relacionadas con la estructura de las proteínas en términos más generales.
Pauling empleó una metodología que le había sido bastante útil para el estudio de la estructura de los materiales inorgánicos como los cristales, hablamos de la difracción por rayos X. Sin embargo, las proteínas mostraron ser menos amenas a los rayos X que los cristales minerales. Este problema metodológico lo fastidiaría por al menos 11 años más. Sin embargo, su empeño se vería recompensado, en 1951 Pauling junto con Robert Corey “1897-1971” y Herman Branson “1914-1995” propusieron correctamente uno de los fundamentos más básicos de la biología molecular, la estructura secundaria de las proteínas, nos referimos a la alfa-hélice y a las beta-láminas. Pauling al igual que la mayoría de la comunidad científica pensaba que eran super-proteínas nucleares y no el ADN las moléculas encargadas de almacenarla información genética, pero aun así realizó una propuesta del modelo de la molécula del ADN (YouTube), su propuesta se puede resumir en la expresión, Triple Hélice. En el modelo de la triple hélice los grupos fosfato altamente negativos están en el centro del cilindro, lo cual hubiera causado una gran tensión electrostática “dos polos negativos no se llevan cuando están juntos”, es por esto que el modelo de Pauling requería grupos fosfato neutralizados, lo cual va en contra de las propiedades ácidas del ADN.
El modelo de Pauling poseía varios conflictos con algunas observaciones experimentales de propiedades del ADN, un ejemplo es que en su estructura de Triple Hélice asumía que los grupos fosfato eran neutrales, lo cual entraba en conflicto con la naturaleza ácida del ADN. Sin embargo, a pesar de esto, otros grupos de investigación competidores se alarmaron sumamente cuando les llegó la noticia de que Pauling estaba trabajando en un modelo para la estructura del ADN, Sir Lawrence Bragg “1890-1971” uno de los líderes del grupo de investigación del laboratorio Cavendish había quedado decepcionado al perder la carrera por el descubrimiento de las estructuras secundarias de las proteínas. Por lo anterior le proporcionó un apoyó fundamental a los trabajos de James Watson “1928-vivo” y Francis Crick “1916-2004”. Este apoyo probablemente les permitió beneficiarse de los datos aun no publicados de Maurice Wilkins “1916-2004” and Rosalind Franklin “1920-1958” del grupo de investigación del King's College que mostraban evidencia de una hélice y una estructura plana uniéndose a lo largo del eje de la hélice.
A principios de 1953 Watson y Crick propusieron una estructura del Ad que se amoldaba de forma más parsimoniosa a los mejores datos que se tenían para la época, la cual sigue en vigencia actualmente y es popularmente conocida como la doble hélice. Tiempo más tarde Pauling, reconociendo que había perdido la carrera por lo que “tan solo un año antes” ahora se conocía era el verdadero fundamento químico de la herencia, citó varios de los errores y limitaciones que tuvo al formular su modelo de la triple hélice. Muchos historiadores de la ciencia están de acuerdo que el mayor problema de Pauling fue no tener acceso a las imágenes de difracción de rayos x logradas por el grupo de investigación de Rosalind Franklin, las cuales eran sin duda las mejores imágenes del ADN disponibles en su tiempo. Más allá de las acusaciones que se puedan realizar a Watson y Crick, su gran éxito se basó en preguntar, en obtener resultados de otros grupos de investigación, cosa que no tuvo Pauling, especialmente en los tan relevantes resultados del grupo de investigación de Rosalind Franklin.

Figura 5.2. Rosalind Elsie Franklin (25 de julio de 1920-Londres, 16 de abril de 1958). Fue una química y cristalógrafa inglesa, responsable de importantes contribuciones a la comprensión de la estructura del ADN (las imágenes por difracción de rayos X que revelaron la forma de doble hélice de esta molécula son de su autoría), del ARN, de los virus, del carbón y del grafito.1 Sus trabajos acerca del carbón y de los virus fueron apreciados en vida, mientras que su contribución personal a los estudios relacionados con el ADN, que tuvo un profundo impacto en los avances científicos de la genética, no se reconoció de la misma manera que los trabajos de James Dewey Watson, de Francis Crick y de Maurice Wilkins.
Pauling no tuvo acceso de forma directa a estos resultados para formular su fallido modelo de la triple hélice, aunque su asistente Robert Corey sí estuvo en contacto, aunque sea con algunos de ellos mientras estaba en una conferencia sobre la estructura de las proteínas en el verano de 1952 en Inglaterra. Pauling no pudo asistir debido a que su pasaporte había sido confiscado por razones políticas, él era un activista político de izquierda, lo cual en el ambiente paranoico de la guerra fría era considerado como comunismo, esta era fue conocida como el periodo McCarthy. Sin embargo, varios autores desestiman la influencia de este inconveniente, ya que Pauling recobró su pasaporte unas semanas más tarde, y pudo viajar a Inglaterra visitando varios grupos de investigación, excepto aquel que lo hubiera llevado a una fama inmortal más allá de la que ya había adquirido, el grupo de investigación en el que trabajaba Rosalind Franklin.
Rosalind Franklin
Uno de los muchísimos aportes de Rosalind Franklin “1920-1958” al modelo de la doble hélice fue su seminario en noviembre de 1951, donde ella presentó dos formas de la molécula, la tipo A y la tipo B, donde demostraba que la posición de los fosfatos debía estar localizada en la parte externa del cilindro. Entre los asistentes a este evento se encontraban James Watson “1928-vivo”. Adicionalmente, ella también especificó la cantidad mínima de agua que le daba estabilidad a la molécula, datos de considerable interés para cualquiera involucrado en el problema de generar un modelo estructural de la molécula del ADN. Otras contribuciones incluyen las mejores fotografías de la época sobre difracciones de rayos X de loa forma B del ADN conocida como la fotografía 51, la cual fue mostrada brevemente a James Watson por Maurice Wilkins “1916-2004” en enero de 1953, y un reporte escrito por un comité de biofísicos que coordinaba al King College llamado Medical Research Council “MCR” fue enviado al director de los laboratorios Cavendish, el doctor Max Perutz “1914-2002” bajo cuya autoridad trabajaban Crick y Watson.

Figura 5.3. La fotografía 51. La famosa foto 51 obtenida por Rosalind Franklin, y que fue la piedra Rossetta para que la estructura del ADN pudiera ser representada por un modelo molecular.
Este reporte del MCR contenía datos del grupo de investigación del King College incluyendo algunos de los trabajos de Rosalind Franklin "1920-1958" y Raymond Gosling "1926-vivo", el cual fue entregado a Francis Crick, quien en ese momento trabajaba en una tesis sobre la estructura de la hemoglobina. La carta fue entregada directamente por Max Perutz. Uno de los aspectos más oscuros de toda esta historia es la ruta de la fotografía 51 aparentemente franklin le había entregado la fotografía a uno de sus alumnos de doctorado llamado Raymond Gosling debido a que ella se encontraba trasladando a otro centro de investigación. Aparentemente Gosling le entregó la carta a Maurice Wilkins quién a su vez se la entregó a Watson. Wilkins, Gosling y Franklin trabajaban en el mismo grupo de investigación, por lo que la transferencia de datos entre ellos es algo bastante normal, lo que no fue normal fue que la información fuese enviada a otro grupo de investigación sin el consentimiento de Franklin. De hecho, la carta del MRC era anormal en sí misma, la tradición inglesa establece que todo documento oficial debe ser secreto hasta que se realiza su publicación ante la comunidad científica, por lo que, los datos aun no publicados de Franklin y Gosling no debieron ser transferidos a otro grupo de investigación antes de su publicación. A pesar de esto Max Perutz no vio ningún daño en mostrarlos de todas formas a Crick.
El trabajo de Rosalind Franklin no fue reconocido en su tiempo, lo cual es una verdadera vergüenza para la comunidad científica de la época, sin su trabajo ningún grupo de investigación hubiera solucionado el problema de la estructura del ADN con la suficiente rapidez, como lo atestigua el hecho de que, sin sus datos, Pauling no fue capaz de dar con la respuesta correcta. El trabajo de Franklin solo comenzó a ser tomado en cuenta con el paso de las décadas, y con orgullo debemos decir que en la actualidad los expertos en la materia le atribuyen su justo lugar en la historia del ADN, sin embargo, aún queda un último peldaño por completar. Los docentes debemos hacer ver que las contribuciones de esta gran mujer fueron cruciales para el establecimiento del modelo de la doble hélice, demostrando que, esos “otros” en la historia de la ciencia son relevantes en su construcción, más aún que los héroes tradicionales que son trasferidos a los libros de texto.
6. La estructura del ADN
6. La estructura del ADN
A representación de las moléculas en espacios escolares es un verdadero dolor de cabeza, esto es porque esas cosas son estructuras que ocupan lugares en el espacio, es decir poseen volúmenes y una estructura tridimensional, lamentablemente la mayor parte de los profesores del planeta solo contamos con un tablero plano y algo con que escribir. Aun con pantallas, la tridimensionalidad mostrada es simplemente una sobra de proyección en perspectiva, es decir, para demostrar las verdaderas formas tridimensionales se requeriría de una suerte de proyector holográfico que muestre la disposición de la molécula, o si no, la creación de modelos en diferentes materiales.

Figura 6.1. James Dewey Watson (Chicago, 6 de abril de 1928-Vivo) Es un biólogo estadounidense, famoso por ser uno de los cuatro descubridores de la estructura molecular del ADN en 1953, junto con el biofísico británico Francis Crick, con el físico Maurice Wilkins y con la química Rosalind Franklin, lo que le valió el reconocimiento de la comunidad científica a través del Premio Nobel en Fisiología o Medicina.
La estructura tridimensional de las moléculas, en especial, de las moléculas orgánicas es de vital importancia para poder entender sus propiedades. Un ejemplo es precisamente las proteínas, ellas actúan mediante sus estructuras tridimensionales. La carrera por entender la estructura de las proteínas fue y aun es una carrera que trae grandes niveles de prestigio a nivel académico. Para el caso del ADN el entendimiento de dicha estructura tridimensional se basó en lo que se conocía de sus componentes químicos, sus propiedades macroscópicas, pero más que cualquier otra cosa, en proyecciones planas obtenidas por el método de difracción de rayos X, mejorado en el King College de Londres por el equipo de investigación de Rosalind Franklin. Sin dichas proyecciones, ningún modelo del ADN se hubiera podido construir con eficacia. A continuación, enumeraremos varios de los componentes del modelo generado pro Watson y Crick empleando la información de Franklin.

Figura 6.2. Francis Harry Compton Crick, OM, FRS (8 de junio de 1916-28 de julio de 2004). Fue un físico, biólogo molecular y neurocientífico británico, conocido sobre todo por ser uno de los cuatro descubridores de la estructura molecular del ADN en 1953, junto con James Dewey Watson y Rosalind Franklin. Recibió, junto a James Dewey Watson y Maurice Wilkins el Premio Nobel de Medicina en 1962 "por sus descubrimientos concernientes a la estructura molecular de los ácidos desoxirribonucleicos (ADN) y su importancia para la transferencia de información en la materia viva".
Las moléculas están compuestas por dos hebras de nucleótidos polimerizados a través del enlace fosfodiester (YouTube; YouTube). Las dos cadenas se organizan en una espiral de torque derecho paralelo, un observador que mira dentro del cilindro, es decir mirando hacia el eje, vería como si las espirales se alejaran siguiendo un giro de manecillas de reloj. La naturaleza helicoidal de la molécula fue uno de las tantas conclusiones que se basaron en la fotografía 51 obtenida por el grupo de Franklin y mostrada a Watson sin su consentimiento. Las dos cadenas son antiparalelas componiendo una doble hélice en la cual cada hebra discurre en una dirección opuesta.

Figura 6.3. Estructura desplegada del ADN. La forma de escalaera lineal solo es verdadera cuando el ADN se despliega, ya sea para replicarse, o para permitir su expresión en forma de mensajes de ARN.
El esqueleto de la cada hebra compuesto por el azúcar y los enlaces fosfodiester de los grupos fosfato se localizan hacia afuera del cilindro, lo cual le permite a la célula diluirse, mientras que las bases nitrogenadas se ubican hacia el interior del cilindro, lo cual les permite interactuar unas con otras por medio de puentes de hidrógeno. Las bases ocupan planos que son aproximadamente perpendiculares a lo largo del eje como los escalones de una escalera en espiral. A parte de los puentes de hidrógeno, las fuerzas de Van der Waals ayudan a estabilizar la molécula de ADN debido a su gran tamaño.
Las fuerzas más importantes son los puentes de hidrógeno que se establecen entre las bases nitrogenadas complementarias. Los puentes de hidrógeno son fácilmente afectados por las fuerzas fisicoquímicas, lo cual le confiere al ADN una naturaleza dinámica en la que puede separarse y recomponerse con relativa facilidad con la ayuda de enzimas especializadas. La distancia entre los grupos fosfato en el exterior del cilindro hasta el centro del cilindro es de 1 nanómetro, por lo que el diámetro del cilindro de la molécula del ADN es de 2 nanómetros. Una pimirimidina en una hebra siempre interactúa con un solo tipo de purina en la hebra antiparalela. Esto implica que la información genética de una hebra es la misma almacenada en su antiparalela, pero codificada en otras bases nitrogenadas.
Los átomos de nitrógeno que se encuentran unidos al carbono 4 de la citosina y al carbono 6 de la adenina se encuentran en la configuración amino y no en la configuración imino. De forma semejante, los átomos de oxígeno unidos al carbono 6 de la guanina y al carbono 4 de la timina se encuentran en la configuración ceto más que en la enol. Estas restricciones estructurales en la configuración de las bases nitrogenadas sugieren que la adenina es la única purina estructuralmente capaz de unirse a la timina, y que la guanina es la única purina capaz de unirse a la citosina, explicando así las reglas de Chargaff a un nivel de la teoría estructural de la química orgánica.
Este modelo se ajustaba perfectamente con la información obtenida por los experimentos de Chargaff. Sin embargo, no existe restricción geométrica en cada peldaño-pareja A-T, C-G, lo cual a su vez nos implica dos cosas. La primera es que la secuencia de nucleótidos no tiene restricciones y por lo tanto, linealmente es tan diversa como una cadena lineal de aminoácidos. La segunda, relacionada con la anterior, es que la molécula de ADN podía tener una complejidad semejante a las de las proteínas, permitiendo así almacenar la información genética. La espiral tiene una configuración externa demarcada por un bucle mayor y un bucle menor, esta configuración le permite a las proteínas estructurales que regulan al ADN reconocer regiones específicas del ADN para diferentes propósitos. La doble hélice realiza un giro completo cada 10 residuos, con un largo aproximado de 3,4 nanómetros.
Funciones del ADN explicadas por el modelo de doble hélice
Cuando los biólogos consideraban las funciones teóricas de la naturaleza química del gen, sugirieron tres funciones básicas que debería asumir dicha molécula:
Almacenar la información genética.
Los genes son entidades químicas que almacenar la información genética, la cual determina los rasgos de los seres vivos. Esta conclusión había sido arrojada por la teoría cromosómica de la herencia y reforzada por las conclusiones de los genetistas postmendelianos comenzando por T. H. Morgan.

Figura 6.4. Microfotografía de ADN bacteriano. A la izquierda tenemos una molécula de ADN bacteriano sin enroscarse, y a la derecha tenemos un ADN del mismo peso molecular, pero enroscado.
Replicación y herencia.
La molécula debe ser capaz de duplicar su concentración durante la fase de síntesis del ciclo celular, de forma tal que explique cómo al finalizar la mitosis se posee un cromosoma simple, mientras que al iniciar la siguiente mitosis aparece duplicado. Esta duplicación permite explicar la multiplicación de los seres vivos.
El artículo original de Watson y Crick no solo exponía una propuesta para el modelo del ADN, también poseía una propuesta sobre cómo es que la molécula podía duplicarse. Watson y Crick propusieron que, durante la replicación, los puentes de hidrógeno que mantienen unidas las dos hebras de ADN se rompen, causando una separación gradual de la hélice. Cada una de las hebras expuesta podría entonces servir como el templado para que nucleótidos complementarios a los de la hebra sean posicionados por una enzima especializada, construyendo de esta forma dos moléculas de ADN a partir de una inicial. Este tipo de replicación es entonces semiconservativo, ya que al finalizar la replicación las dos moléculas de ADN poseen una hebra de la generación anterior y una hebra nueva. Este es un excelente ejemplo de predicción, ya que pasarían varios años hasta que esta propuesta fuera comprobada experimentalmente.
Expresión de la información genética
Durante el tiempo en que se investigó al ADN otra línea de investigación conllevó a determinar que los genes gobernaban la formación de moléculas biológicas llamadas proteínas, las cuales se encargan de generar los rasgos biológicos. Cualquiera que fuera la naturaleza química de los genes debía cumplir con estas condiciones.
De las tres funciones, la que relaciona al ADN y su estructura con las proteínas permanecería oscura al menos por una década más, pues deberían describirse la función del otro ácido nucleico, el ARN. Sin embargo, la importancia de proponer un modelo para la estructura del ADN fue enorme, ya que no solo explicaba la mayor parte de la información sobre la época en una gran gama de líneas de investigación, también planteó nuevas preguntas, por lo que señalaba nuevas líneas de investigación. Una vez que el modelo fue aceptado por la mayor parte de la comunidad científica, toda teorización sobre la herencia y la evolución debería tener en cuenta su estructura.

Figura 6.5. Super-estructura del ADN. La consecuencia del giro por debajo "underwound" o por encima "overwound" afectan el modo en que la molécula adquiere bucles distintivos, el giro por debajo es más permisivo con la replicación y la expresión del ADN.
Super-estructura del ADN
En 1063, Jerome Vinograd y sus colegas del Caltec encontraron dos moléculas de ADN cerradas y circulares de idéntica masa molecular podían exhibir tasas de sedimentación muy diferentes durante la centrifugación. Análisis posteriores indicaron que la molécula de ADN se sedimentaba as rápido en la medida que la apariencia de la molécula es más compacta, es decir, presenta un aspecto enrollado sobre sí misma. Para la época las microfotografías de electrones ya podían mostrar las moléculas de ADN, en la figua1 puede notarse como en la parte (a) se presenta una molécula de ADN con un aspecto relajado, con pocos nudos, mientras que en (b) se encuentra enrollada. Cuando la mezcla de ADN relajado y superenrollado son sometidos a electroforesis (YouTube) se generan dos bandas de corrimiento, el ADN enrollado avanza más que el ADN relajado. Análisis posteriores indicaron que la molécula del ADN se sedimenta más rápidamente cuando su forma esta enroscada, semejante cuando se tiene una bola compacta de hilo d forma muy densa.

Figura 6.6. Super-enrollamiento. A diferencia de las luces de navidad, para que el ADN pueda realizar cualquiera de sus funciones, ya sea la replicación o la expresión de sus genes, requiere de que la topología cambie de una entidad compacta a una fibra elongada, donde incluso la Doble Hélice se desenrosque, exponiendo cada hebra de ADN.
El superenrollamiento es un término muy complejo, pero no es exclusivo ni de las moléculas o del ADN, es más, cualquiera que haya lidiado con cuerdas lo habrá enfrentado más de una vez. Yo mismo puedo poner como ejemplo los primeros días del mes de navidad cuando sacas de la gran caja las luces de navidad “que por cierto son una gran analogía al genoma en otras cosas”. Cuando sacas las luces se encuentran haciendo nudos inextricables difíciles de desenredar, a esa estructura en topologías se lo llama superenrollamiento, y si, posee una descripción matemática muy precisa, aunque eso de poco nos sirve a quienes nos toca desenredar esa masa informe y apretujada. Del mismo modo el genoma puede apretarse, pero para compactarlo o desenredarlo no cuenta con manos inteligentes, en el mundo molecular la molécula de ADN depende de enzimas que se encargan de enredar, desenredar, girar, despegar y pegar hebras de ADN; es decir, se comportan como manos, tijeras y conectores.
El superenrollamiento puede caracterizarse por dos formas dependiendo del tipo de giro que la molécula adopte con respecto al punto de vista arbitrario del observador. En primera instancia el ejemplo típico que se realiza es con una molécula de ADN circular, la cual ya está enrollada en si misma debido a la estructura helicoidal del ADN. Ahora todo depende del observador, que desde el punto de vista de las imágenes se encuentra arriba. Al realizar un giro del círculo se generan dos bucles. Si avanzamos desde el bucle superior a la derecha y la molécula pasa por debajo de su misma, decimos que se ha enroscado por debajo “underwound”. Si por el contrario la molécula pasa por encima de sí misma decimos que se ha enroscado por encima “overwound”.

Figura 6.7. Resolviendo el super-enrollamiento. Las topoisomerasas (YouTube) alteran la topología del ADN, generando o resolviendo los bucles.
Cuando el ADN se encuentra enroscado por debajo de sí mismo se dice que se encuentra en un estado de superenrollamiento negativo, y, por el contrario, cuando está enroscado sobre sí mismo diremos que se trata de un superenrollamiento positivo. El ADN circular de las bacterias, las mitocondrias, los cloroplastos y los virus tienden a enroscarse de forma negativa. Sin embargo, no solo el ADN circular tiende al enroscamiento negativo, virtualmente todos los genomas se enroscan de este modo, y es probable que se deba a limitantes fisiológicos. Lo anterior se debe a que el enroscamiento negativo ejerce una fuerza que ayuda a separar las dos hebras de la Doble Hélice cuando la molécula va a replicarse o a expresar la información genética.
Todos estos cambios del AND ocurren como cambios de su superficie y no de su secuencia, por lo cual su estudio se denomina topologías del ADN. Dado que la célula depende de proteínas especializadas para alterar las topologías, a estas enzimas se las denominan toposiomerasas “alteran la molécula a topologías diferentes sin alterarla molecularmente”. Las topoisomerasas fueron descubiertas experimentalmente en 1971 por James Jang de la Universidad de California, Berkeley. Las células poseen una variedad de topoisomerasas que pueden dividirse en dos clases generales.
Las topoisomerasas del tipo I alteran el estado de superenrollamiento de la molécula de ADN al crear un corte transitorio en una de las hebras del ADN. Posteriormente la molécula gira sobre sí misma para volverse a conectar. Este tipo de giro relaja el superenrollamiento de la molécula. Este tipo de movimiento es vital para los eventos principales del AD denominados replicación y transcripción, pues para lograrlos, la molécula debe relajarse de una forma que quede prácticamente en un estado lineal.
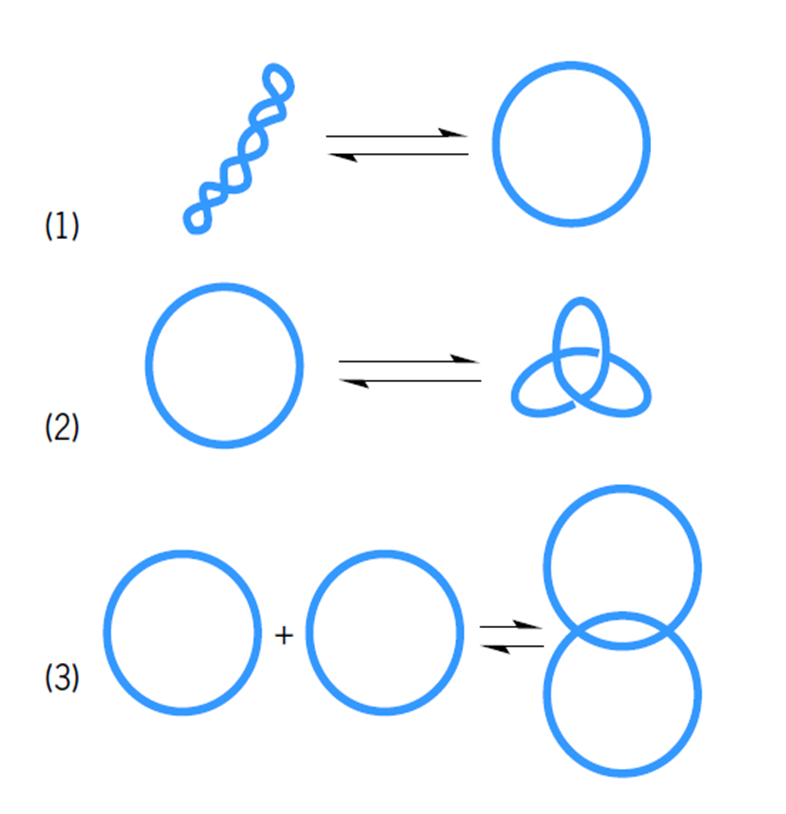
Figura 6.8. El ADN puede enrollarse de diversas maneras. Otras topologías que pueden ser mediadas reversiblemente por las topoisomerasas de tipo II.
El superenrollamiento puede ser algo muy malo, y cualquiera que le haya tocado desenredar las luces de navidad sabrá de lo que estoy hablando. Las moléculas del ADN no se desenredan del modo en que lo hacen las luces de navidad en el sentido de buscar una organización de todo el sistema. Para desenredar nudos que generan alta tensión y por lo tanto obstaculizan la replicación y expresión de los genes han evolucionado las topoisomerasas de tipo II. Una topoisomerasa de tipo II interactúa con dos moléculas que se han enroscado y generan gran tención. Cuando se ubica una zona de tención, la topoisomerasa corta una de las moléculas, dejando a la otra pasar derecho, aliviando la presión y desenredando el sistema en el proceso. Evidentemente puede revertir el proceso para enroscar dos moléculas de ADN. Adicional a esto, las topoisomerasas del tipo II pueden encadenar varias moléculas circulares de ADN en estructuras complejas y compactas.
7. La replicación del ADN 1, el experimento
7. La replicación del ADN 1, el experimento
La reproducción es una de las propiedades más importantes de los seres vivos, a tal punto que no existe definición de ser vivo que no la toma en consideración como una de las propiedades sine qua non que debe poseer cualquier sistema para ser designado como vivo. Inicialmente el proceso de reproducción es estudiado a nivel estructural y biológico, sin embargo, durante la etapa de síntesis en el ciclo celular se da otra historia, una historia de índole bioquímico. La célula duplica la concentración de ADN en el núcleo empleando una serie de proteínas especializadas, las cuales también están involucradas en la reparación del genoma, lo cual reduce al máximo la cantidad de mutaciones posibles, pero no las elimina por completo.

Figura 7.1. Replicación del ADN. Acercamiento a la propuesta original para la replicación del ADN, cada una de las hebras antiguas "azul" sirven como templado para la creación de la nueva "anaranjado", haciendo del proceso algo semiconservativo.
El modelo propuesto por Watson y Crick basado en las difracciones de rayos X realizadas por Rosalind Franklin en 1953 fue acompañada por una sugerencia para un mecanismo de replicación autodirigido. Las dos hebras de la molécula se mantienen juntas mediante puentes de hidrógeno generados por las bases nitrogenadas, dos para la pareja adenina-timina y tres para la pareja citosina-guanina. El rompimiento de estos puentes de hidrógeno podía realizarse por temperatura in vitro, pero in vivo debía involucrar una batería de enzimas especializadas. Una vez que las dos hebras estuvieran separadas, cada una serviría como patrón o templado para la formación de una hebra nueva.
El modelo de Watson y Crick realiza ciertas predicciones muy específicas con respecto al comportamiento del ADN durante su replicación en la fase de síntesis del ciclo celular. De acuerdo con su propuesta, cada una de las moléculas hijas posee una hebra madre original heredada y una hebra hija formada a partir de la hebra madre que sirve como templado. A este tipo de replicación se la denominaría semiconservativa. Evidentemente esta no era la única hipótesis en su tiempo a cerca de la replicación del ADN.
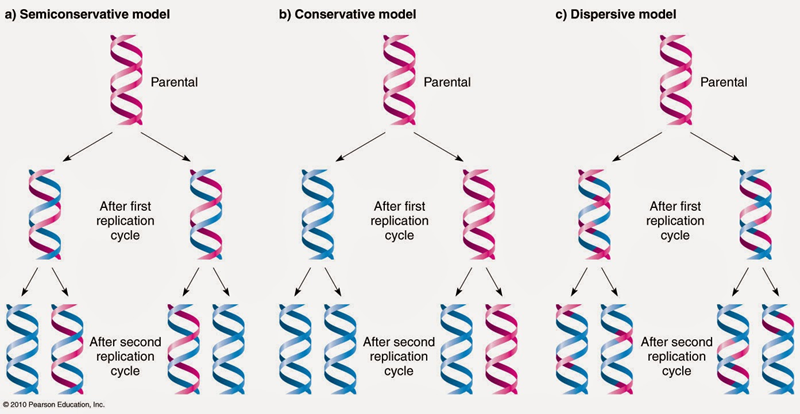
Figura 7.2. Hipótesis para la replicación del ADN. "Izquierda" modelo de replicación semiconservativa durante dos generaciones. "Medio" modelo de replicación conservativa durante dos generaciones. "Derecha" modelo de replicación disruptiva durante dos generaciones.
En la replicación conservativa, las dos hebras originales se volverían a juntar, de modo tal que crearía dos hebras hijas que a su vez se juntaría, de este modo se crearía una molécula de doble hélice hija y una molécula de doble hélice madre. En la hipótesis de la replicación dispersa, las hebras de la molécula madre se romperían en fragmentos, y la nueva molécula se sintetizaría a partir de estos fragmentos cortos. Los fragmentos viejos y los nuevos serían entonces reensamblados al azar, generando así una mezcla de ADN parental y ADN recientemente sintetizado.

Figura 7.3. Marca radioactiva/masa de nitrógeno. Los símbolos y colores arbitrarios para la cadena pesada "izquierda" y ligera "derecha" asociadas al nitrógeno 15 y nitrógeno 14 respectivamente. Dado que el marcador es el nitrógeno, la diferencia entre las dos hebras de ADN se ubica en las bases nitrogenadas
Aunque la replicación disruptiva se veía como poco probable, la única forma de refutar los modelos era poder distinguir entre las secuencias madre y loas secuencias hijas en una replicación controlada. El método para lograr esto fue desarrollado en 1957 por Matthew Meselson "1930-vivo" y Franklin Stahl "1929-vivo" del Caltec empleando isótopos pesados de nitrógeno 15 e isótopos ligeros de nitrógeno 14. Los diferentes isótopos ayudarían a distinguir entre las hebras madre e hija. El principio es la densidad, una molécula que contiene únicamente nitrógeno 15 es más densa, mientras que otra que contiene solo nitrógeno 14 será menos densa. La relación de densidad intermedia será de números fijos en el caso de una replicación semiconservativa, o de una variación continua entre los dos extremos en el caso de una replicación disruptiva al azar.

Figura 7.4. Efecto macroscópico en centrífuga. Al centrifugar los blancos se obtienen dos fases "invisibles", una para las moléculas densas y otra para las moléculas ligeras. La centrifugación es un proceso que sirve para separar sustancias por su densidad, en el ejemplo de la sangre, los glóbulos rojos tienden al fondo por su alto contenido de hierro, en medio quedan los glóbulos blancos, y en la parte superior queda la solución líquida llamada plasma. El proceso funciona también para el ADN (YouTube).
El experimento Meselson-Stahl sobre la replicación del ADN
Este experimento será uno de los experimentos cruciales en la historia de la biología molecular. Sus resultados permitirían corroborar que la propuesta de replicación generada por Watson y Crick junto con su modelo de la doble hélice era correcta. Hay que rescatar que los biólogos moleculares no poseen microcamaritas para ver el genoma, por lo que en su contexto cualquier hipótesis sobre la replicación del ADN podría ser correcta. Las hipótesis a manejar eran tres: replicación semiconservativa, replicación conservativa y replicación disruptiva. La metodología empleada fue mediante isótopos marcados, la medición se realizaría por la densidad de la molécula resultante.
ADN patrón
Primero que todo hay que crear el patrón para la molécula de ADN de alta densidad, para esto simplemente se hace crecer una colonia de bacterias en un medio que solo contiene nitrógeno 15. Con el tiempo, la colonia solo puede integrar nitrógeno 15 en su nuevo ADN. De esta muestra se toma una parte para la determinación de la densidad y otra para realizar la fase experimental. Por otra parte, se tendría un cultivo de bacterias que crecen en un medio que contiene solo nitrógeno 14, este cultivo proporcionará la muestra para determinar la densidad de una molécula de ADN que contiene solo nitrógeno 14. Con los valores de los extremos, muy denso y poco denso puede crearse una escala con las muestras experimentales en los siguientes pasos.
La determinación de la densidad fue realizada mediante la centrifugación de equilibrio o centrifugación isopicnica. En este procedimiento la muestra de ADN sometida a la centrifugación se mueve de forma diferencial en el tubo. La más densa termina en el fondo y la más ligera termina cerca de la superficie. En el experimento Meselson-Stahl, el blanco de ADN con nitrógeno pesado termino más al fondo, mientras que la muestra que había sido incubada con medio de nitrógeno ligero terminó más cerca de la superficie. En medio de ambas se pudo crear una escala, entre más cercana fuera la molécula a la forma perfectamente pesada, esta migraría al fondo, y entre más cercana fuera a la forma perfectamente ligera esta quedaría cerca de la superficie.
Espectrofotometría
Bueno, usted centrifuga, pero lo que sale de la centrífuga es un fluido transparente, el problema es ver lo que es invisible al ojo humano. Para esto, se pasa el fluido del tubo de centrifuga a otros tubos en orden como se muestra en Figura 7.5.

Figura 7.5. Experimento de Meselson-Stahl. (A) Cada uno de los blancos arrojan picos únicos en los extremos de la tabla.
Para observar lo que no puede ser visible se emplea el método de espectrofotometría, la cual arroja picos dependiendo del tubo de muestra que has obtenido al decantar el tubo de centrifugación. De esta forma cada una de las muestras puras arrojará picos semejantes a estos Figura 7.6.

Figura 7.6. Experimento de Meselson-Stahl. (B) La espectrofotometría arroja valores con los cuales se puede construir una gráfica que indica la cantidad de sustancia en una solución. Al decantar el tubo centrifugado en varios tubos, estos generan un gradiente de concentraciones. A estos tubos se les extraen muestras para el espectrofotómetro y así obtener las concentraciones
Resultados esperados vs experimentales
En el modelo conservativo se esperaría la formación de dos bandas al final del procedimiento, una para las hebras pesadas y otra para las hebras ligeras. Cada pico corresponde a las hebras originales con las que inició el cultivo y a las hebras hijas que son completamente nuevas y ligeras Figura 7.7.

Figura 7.7. Experimento de Meselson-Stahl. (C) Este es el resultado más cercano, dos picos uno para las moléculas conservadas y otro para las nuevas, sin intermedios de ninguna clase.
El modelo disruptivo propone la destrucción de las cadenas de la primera generación y la conformación de moléculas hibridas en fragmentos, con el paso de las generaciones las moléculas integraran más y más del nitrógeno ligero. En el modelo semiconservativo durante la primera generación, todas las moléculas pesadas son convertidas a moléculas híbridas, y en las siguientes generaciones, aunque se producen más moléculas ligeras, siempre se conserva una cantidad de moléculas hibridas. Los resultados del experimento arrojaron la formación del pico esperado para la molécula híbrida, es decir, corroboraron los resultados esperables si la molécula realizara la replicación de forma semiconservativa. Para 1960 pudo demostrarse lo mismo en el modelo biológico de los eucariotas, por lo que, en el ser humano, la molécula de ADN también se realiza la replicación de ADN de forma semiconservativa. El procedimiento experimental involucraría el uso de un marcador medible mediante microfotografía de los cromosomas.
8. La replicación del ADN 2, en bacterias
8. La replicación del ADN 2, en bacterias
Aunque suene un poco antropocéntrico, en términos cualitativos y cuantitativos, el ADN de las bacterias se encuentra organizado de un modo más sencillo que en los eucariotas, por esta razón fue primero en las bacterias donde se pudo estudiar el modo en que el ADN se replica. Específicamente, el modelo biológico fue Escherichia coli, la bacteria más conocida por la ciencia y de quien es posible que se tenga una leyenda negra. Igual que sucede con la mayoría de las bacterias, E. coli posee una enorme diversidad de variantes a las cuales llamamos cepas, algunas cepas son muy peligrosas e incluso hasta fatales como E. coli O157H7, pero las cepas que se emplean en el laboratorio de biología molecular son inocuas. Y no es que una bacteria inocua genere por arte de magia todos los mecanismos de patogenicidad necesarios para hacerse infecciosa en unas cuantas generaciones, el empleo de E. coli es completamente normal y seguro en laboratorios de bajos niveles de bioseguridad.
Figura 8.1. Cultivo bacteriano. A una temperatura restrictiva un mutante termosensible no podría generar colonias como las que se ven en la imagen, aunque la bacteria esté allí no puede desarrollarse.
Dos aproximaciones experimentales serían cruciales para el entendimiento de la replicación del ADN en las bacterias, la primera es el empleo de mutantes sensibles y la otra es el de la replicación in vitro. La existencia de cepas de laboratorio con mutaciones que impiden la síntesis de una o varias de las enzimas necesarias para la replicación. Aunque esto pueda parecer extraño, el aislamiento de mutantes su cromosoma solo es posible si la mutación es sensible a la temperatura, lo cual permite el majeo de ambos fenotipos.

Figura 8.2. Cultivando bacterias. En este contexto, in vitro se refiere a que las proteínas y los procesos se llevan a cabo fuera de las células, mientras que in vitro se refiere al proceso que se lleva a cabo al interior de la célula.
Cuando necesitas hacer crecer el cultivo lo haces a una temperatura permisiva, y cuando requieres ver el efecto de la mutación, haces crecer el cultivo a una temperatura restrictiva. Los mutantes sensibles a la temperatura han sido aislados para virtualmente cualquier actividad fisiológica, y han sido particularmente importantes para el estudio de la síntesis de ADN durante la replicación, la reparación y la recombinación genética.

Figura 8.3. Pinzas de replicación. Las pinzas de replicación en un cromosoma bacteriano, abren el ADN para que las proteínas asociadas inicien la copia y síntesis de nuevo ADN.
El desarrollo de sistemas in vitro en el que la replicación puede ser estudiada empleando componentes extraídos y purificados de la célula. En algunos estudios la molécula de ADN es incubada con extractos celulares en los que las proteínas sospechosas de tener algo que ver con la replicación son empleadas o removidas, confirmando resultados con los mutantes termosensibles. Tomados en conjunto, estas aproximaciones experimentales han revelado la actividad de más de 30 proteínas diferentes necesarias para la replicación del ADN en Escherichia coli. En los siguientes artículos discutiremos estos procesos en la medida en que han sido aclarados algunos, pero otros aún siguen en investigación.
Aunque es totalmente merecido que se designe a la molécula del ADN como la molécula de la vida, si es necesario recalcar que, por sí mismo el ADN está tan vivo como cualquier polímero plástico sintético que se convierte en fibras transparentes en un vaso de precipitado. El ADN por sí solo, desnudo no está vivo, no puede realizar ninguna de las funciones que asociamos con la vida, es necesaria la presencia de una enorme batería de proteínas, es decir, de la célula, para que este pueda replicar, producir y mutar. Esta aparente dicotomía entre la función del ADN y la función de las proteínas conllevaría a la definición del problema del huevo y la gallina en el origen de la vida. Sin embargo, para nuestro contexto, no discutiremos aun este problema.
Pinzas de replicación
El cromosoma de las bacterias es circular, esto condiciona mucho de la maquinaria molecular empleada para replicarlo. La replicación inicia en un sitio específico del cromosoma bacteriano, este lugar se denomina origen de replicación. El origen de replicación en Escherichia coli es una secuencia específica llamada oriC donde varias proteínas se ensamblan para iniciar el proceso de replicación. Una vez iniciado, el proceso se da en ambas direcciones opuestas, lo cual se conoce como bidirecionalidad. Los sitios donde los segmentos replicados se unen con el ADN no replicado se denominan pinzas de replicación. Cada pinza de replicación corresponde al sitio donde la doble hélice parental está siendo separada y donde los nucleótidos están siendo incorporados.

Figura 8.4. Replicación de un cromosoma bacteriano. Cada una de las pinzas de replicación sintetiza ADN, para que el cromosoma se duplique, el proceso es tan rápido que no se ha terminado de completar el cromosoma nuevo cuando ya inicia su propia replicación.
Las dos pinzas de replicación se mueven en direcciones opuestas hasta que se encuentran en el punto opuesto del origen de replicación. En este punto, los dos cromosomas se separan y migran a los polos opuestos de las células. Es importante resaltar que debido a la peculiaridad del modo de vida de Escherichia coli, la replicación del cromosoma dará inicio aun antes de que las dos células hijas se hallan separado. Esto hace de la replicación bacteriana algo muy rápido, ya que la fase de síntesis y de crecimiento se da de forma simultánea.
Relajación y separación de las hebras
Para que el ADN pueda ser replicado o leído, las hebras de ADN deben estar relajadas y liberadas en una configuración simple y lineal. Cuando las dos hebras de la doble hélice de un cromosoma circular empiezan a separarse ocurre algo que es muy común para cualquiera que trata de lidiar con cuerdas o fibras y es el superenrollamiento. En resumen, la separación de las dos hebras empieza a generar una tensión de torsión en las zonas del cromosoma que aún no han sido separado generando un superenrollamiento positivo. A esto se lo denomina relajación “unwinding” de la doble hélice.
A diferencia de una cuerda que puede superenrollarse, el ruperenrollamiento del ADN provocaría la obstrucción de la replicación a medida que las pinzas de replicación se aproximan a los nudos de mayor tensión. Considerando que un cromosoma bacteriano de Escherichia coli puede generar en potencia 400 000 nudos, y que estos deben ser resueltos en menos de 40 minutos, la magnitud de este problema hace ver al nudo gordiano como un juego de niños. ¿Cómo resolver este problema topológico en un marco de tiempo entre minutos y segundos? La respuesta es el empleo de las topoisomerasas de tipo II.

Figura 8.5. Dirección de replicación. Originalmente se proponía que ambas hebras crecían de forma continua en dirección a la pinza de replicación.
Las enzimas especializadas en aliviar las tenciones de torsión en el superenrollamiento del ADN son las tipoisomerasas de tipo II, específicamente para la replicación, la enzima es denominada ADN girasa, la cual como cualquier topoisomerasa de tipo II corta el ADN permitiendo a la hebra que genera la tención girar, luego esta misma enzima repara la hebra cortada. La ADN girasa viaja a lo largo de la cabeza de la pinza de replicación, removiendo los nudos “superenrollamientos positivos”, el proceso requiere sacrificar energía en forma de ATP. En los eucariotas el mecanismo es similar.
Las polimerasas de ADN

Figura 8.6. Arthur Kornberg. (Brooklyn, Nueva York; 3 de marzo de 1918-Stanford, California; 26 de octubre de 2007) fue un bioquímico estadounidense. Estudió medicina en la Universidad de Rochester, donde se doctoró en 1941. Permaneció trabajando en el Servicio de Salud Pública de Estados Unidos durante diez años. En 1952 fue nombrado jefe del Departamento de Microbiología de la Universidad Washington en San Luis, posteriormente aceptó la plaza de jefe del Departamento de Bioquímica de la Universidad Stanford, de California.
La molécula de ADN en el modelo aceptado posee dos hebras que son antiparalelas y se designan en base al carbono que ceda libre en el azúcar en cada punta. Si una de las hebras inicia en el extremo 3´ entonces terminará en el extremo 5´. De forma antiparalela la hebra complementaria debe comenzar en el extremo 5´y terminar en el 3´- Los sistemas biológicos tienen una serie de complicaciones extrañas, por ejemplo, uno esperaría que la enzima que duplica al ADN posea dos versiones también antiparalelas, mientras que una cataliza la copia desde 3´→ 5´, la otra copiaría la versión antiparalela desde 5´→3´. Sin embargo, en la realidad esto no es tan fácil.

Figura 8.7. Modelo de peine. Cuando dos peines se reconocen forman la escalera, que es una analogía más común para la estructura del ADN
El estudio de las enzimas que se encargan de copiar al ADN comenzó en la década de los 50s, básicamente cuando la comunidad científica acepto que (1) la molécula de ADN era la naturaleza química del gen y que (2) existiera un modelo para la estructura química de dicha molécula, el problema a resolver fue como es que esta molécula logra copiarse a sí misma. En la misma década en que estos dos conceptos se aceptaron “1950-1960”, Arthur Kornberg “1918-2007” de la Universidad de Washington dio inició al estudio de las enzimas replicativas. En sus estudios iniciales, Kornberg y colaboradores purificaron una enzima de extractos bacterianos, esta enzima era capaz de incorporar nutrientes con elementos marcados radioactivamente, los cuales eran empleados para construir nuevo ADN. Esta enzima fue denominada polimerasa de ADN, aunque años después será renombrada como la ADN polimerasa I.

Figura 8.8. Separación del peine. Cuando cada hebra está separada, cada una forma un peine.
Para que la síntesis de ADN pudiera ser llevada a cabo por la ADN polimerasa I, la enzima requiere de todas las cuatro bases nitrogenadas, de lo contrario la copia se detiene. Esto implica que la enzima agrega de una en una cada base y cuando faltan se detiene en el ensamblado. Si todo sale bien, las hebras marcadas radioactivamente poseían la misma composición en términos de porcentajes de bases nitrogenadas que las secuencias parentales, lo cual sugería que las secuencias madre servían como templado de las secuencias hijas.
Si bien admiramos el modelo de Watson y Crick para el ADN, a la hora de visualizar la función de los genes o como en este caso la replicación, dicho modelo se hace muy complejo. Es por esto que es bastante común en los libros de biología molecular expresar al ADN mediante modelos simplificados. El modelo empleado para representar la molécula de ADN en la replicación y la transcripción es el modelo de peine. Se lo llama así porque el esqueleto de la molécula es representado como una cinta, mientras que las bases nitrogenadas se representan como las cerdas del peine.
Analicemos un poco más sobre esto, dado que la secuencia hija es generada a través del copiado de la secuencia madre, la similitud entre la secuencia de dos moléculas de ADN establece una relación directa de parentesco. Las enzimas no son perfectas y como veremos posteriormente poseen un ritmo de errores, lo cual conlleva a que la molécula que se copia será semejante, pero no exactamente igual a su molécula madre. De lo anterior se concluye que entre menos parentesco tienen dos moléculas de ADN el nivel de similitud entre sus secuencias disminuye cuantitativamente. Este razonamiento será de vital importancia para la genética evolutiva, pues si las especies están relacionadas de forma filial ancestro-descendiente, sus secuencias de ADN deben poseer grandes niveles de similitud entre sí. Esta es una predicción sobre la que volveremos en artículos futuros.

Figura 8.9. Importancia del cebador. Sin cebador la polimerasa de ADN no se acopla al templado para iniciar el copiado.
Como vimos anteriormente, el cromosoma bacteriano se relaja cuando inicia la replicación, esto se debe a que si la ADN polimerasa interactúa con una molécula de doble hebra, no ocurre nada. Esto fue un resultado inesperado para los primeros biólogos moleculares. En conclusión, para que la ADN polimerasa pueda iniciar la replicación es absolutamente necesario que la molécula esté relajada, es decir, que las dos hebras del ADN se encuentren separadas y en una disposición lineal.
Lo que fue verdaderamente extraño es que la ADN polimerasa junto con una molécula de una sola hebra de ADN tampoco hace nada (…), pero cuando se adicionaban fragmentos de ADN muy cortos “unos 20 nucleótidos” a la mezcla, inmediatamente comenzaba el proceso de replicación.
Ni una molécula de doble hebra, ni otra molécula de hebra simple son capaces de iniciar la replicación. Se requiere de una hebra relajada sobre la cual se posiciona un segmento de ADN muy corta, sobre esta punta es que la polimerasa de ADN se posiciona, en el extremo 3´ y comienza a adicionar nucleótidos, alargando la molécula siempre por la punta 3´. Dado que el segmento corto de ADN es el que da el punto de inicio para la copia se lo ha denominado cebador, secuencia iniciadora o primer para la copia del ADN.

Figura 8.10. Copiado dirección 5→3. Acoplado el cebador, la ADN polimerasa inicia la síntesis de la cadena complementaria nueva.
Todas las polimerasas de ADN sean procariotas o eucariotas tienen los mismos requisitos básicos: (1) Una molécula de ADN relajada que exponga cada una de las hebras de forma lineal, esta servirá como el original o templado a partir del cual se generará la copia; (2) Un fragmento cebador o primer de ADN que permite a la polimerasa adicionarse en el extremo 3´ e iniciar la adición de nucleótidos. Estos requisitos explican porque ciertas estructuras de ADN fallan en promover la síntesis del ADN. Una molécula de doble hebra no hace nada porque no expone el templado, una molécula de hebra simple perfecta tampoco hace nada porque carece del primer. Una molécula de hebra simple con pequeños segmentos dobles permite que las polimerasas se peguen en el extremo 3´e inicien el copiado. Sin embargo, el descubrimiento de los cebadores in vitro levantó la pregunta ¿Cómo ocurre el proceso in vivo?
La ADN polimerasa purificada por el grupo de investigación de Kornberg identificó una propiedad limitante en la polimerasa de ADN, la cual sería luego encontrada en las demás. Las polimerasas de ADN solo pueden pegarse en la punta 3´ del primer, y toma a los nucleótidos pegando el extremo 5´, y al avanzar sobre el nucleótido nuevo vuelve a posicionarse en el extremo 3´. Lo anterior implica que las moléculas de ADN se encuentran limitadas a sintetizar solo en la dirección 5´→ 3´.

Figura 8.11. Dirección no permitida. Las polimerasas de ADN son unidireccionales, pueden copiar de 5→3, pero no de 3→5.
En un claro contraste a lo que propusieron originalmente Watson y Crick, no existe versión de la ADN polimerasa que catalice la síntesis de ADN en la dirección 3´→ 5´ lo cual complica el mecanismo de replicación de la hebra de ADN de la misma dirección. En el diagrama de replicación del cromosoma circular se esperaría que ambas hebras se sinteticen de forma continua, esto es fácil para la cadena 5 ´→ 3´, pero la otra hebra presentará dificultades. Por el momento llamaremos a la cadena de fácil replicación como hebra líder o cadena líder, mientras que la cadena de difícil replicación será denominada hebra rezagada o cadena rezagada. Sin embargo, los resultados de Kornberg aún no refutaban la existencia de la hipotética enzima que cataliza en la hebra rezagada de forma antiparalela, en su contexto los biólogos moleculares aún se preguntaban por la existencia de otras polimerasas de ADN, y por la posibilidad de que, en el contexto celular, el proceso ocurriera de una forma diferente.
Durante la década de 1960-1970 se dieron pistas de que la polimerasa de Kronberg no era ni la púnica ni la más importante polimerasa de ADN. En 1969 la pista fundamental para dicha aseveración fue el aislamiento de un mutante de Escherichia coli que tenía una mutación que inhabilitaba la función de la polimerasa de Kronberg en casi un 99% y aun así, la bacteria era capaz de replicarse a una velocidad normal. Estudios posteriores revelaron la existencia de otras poliomerasas de ADN, por lo que la polimerasa de Kronberg fue denominada ADN polimerasa I.
Tiempo después se encontró que la polimerasa que contribuía principalmente a la replicación del ADN fue la tercera en ser descubierta, es decir, la ADN polimerasa III. Una bacteria tipia posee entre 300 y 400 moléculas de ADN polimerasa I pero solo 10 copias de la ADN polimerasa III. En otras palabras, aun cuando su efecto es más potente, pasaba de ser percibida por la ADN polimerasa I. Sin embargo, ninguna polimerasa que se iba descubriendo fue capaz de resolver el enigma, todas se pegaban exclusivamente al extreme 3´de cebador.
Replicación semidiscontínua
El problema con el que se encontraron los biólogos moleculares a la hora de determinar la función de las polimerasas de ADN es que todas sintetizan al pegarse en el extremo 3´de un cebador o primer de ADN. Esto traía consecuencias sobre el modo de replicación propuesto por Watson y Crick que presuponía la existencia de una ADN polimerasa que podía pegarse al extremo 5´y copiar.
los cebadores son secuencias de ADN, pero la pregunta surgía, ¿Cómo podía crearse el cebador si ninguna polimerasa de ADN podía crearlo en primera instancia? Los cebadores al ser ADN requerirían una polimerasa de ADN, pero las polimerasas de ADN solo actúan cuando hay un cebador presente, lo cual nos trae a un dilema de tipo huevo gallina. En la siguiente serie de artículos veremos cómo es que estos dilemas fueron resueltos experimentalmente.
La falta de polimerización de las polimerasas de ADN en la dirección 3´→ 5´posee una explicación bastante simple, es imposible químicamente, eso es todo. Los procesos biológicos son en últimas al nivel moléculas, procesos que dependen de las leyes de la química. Todo está en la química del carbono 5 y el carbono 3 en la desoxirribosa. Ambos carbonos portan el grupo fosfato, pero a diferencia del carbono 5´, el 3´no puede girar, y esta posición facilita un ataque nucleofílico por parte del nucleótido que ingresa a la cadena. Esta estabilidad estructural facilita el reconocimiento de la enzima, debido a que las enzimas reconocen formas, de modo tal que las polimerasas de ADN solo pueden reconocer el carbono estable sobre el cual puede realizarse el ataque nuclefilico.

Figura 8.12. ¿Cómo se replica el ADN en el lado inverso a la síntesis 5→3?. Más que hebra líder deberíamos decirle lado líder, siempre que un primer o cebador expone una punta 3´, la ADN polimerasa puede alargarla en la dirección de abertura de la hebra. Dado que la hebra se abre bidireccionalmente, esto permite que dos lados puedan copiarse continuamente. El problema es lo que ocurre en la dirección 5´ hacia atrás.
La síntesis de ADN es un proceso que gasta enormes cantidades de energía metabólica. Esto se evidencia en la estructura misma de los nucleótidos adicionados, pues se encuentran en la forma trifosfatada. De forma homóloga a la adenosina que al adquirir tres grupos fosfato se convierte en la molécula de mayor energía en los sistemas biológicos, los nucleótidos del ADN deben estar en una forma trifosfatada para poder ser empleados por la ADN polimerasa. Cuando la polimerasa adiciona un nucleótido a la cadena creciente, debe cortar dos de los grupos fosfato para que la reacción pueda realizarse. La adición del nucleótido depende de su capacidad para ser reconocido por la base complementaria del templado, formando puentes de hidrógeno estables, aunque en ocasiones este mecanismo de reconocimiento falla generando una mutación puntual o SNP.

Figura 8.13. Replicación semidiscontínua. Para poder copiar lo que hay atrás del extremo 5´ es necesario adicionar un nuevo cebador o primer y seguir copiando desde la dirección 3´hacia el primer cebador. Esto implica que solo puede copiarse por segmentos a medida que la hebra se abre por el otro lado, adicionando un cebador a la vez y copiando parte por parte.
La polimerización de una cadena de ADN requiere de un primer o cebador, la polimerasa de ADN se coloca en el carbono más estable que posee un grupo fosfato, es decir, el carbono 3´. El modelo simplificado más próximo para explicar la adición de nucleótidos a la cadena creciente se denomina mecanismo de doble ión metálico. Y es aquí donde algo interesante emerge, la reacción de polimerización de ADN no puede llevarse a cabo sin la intervención de iones metálicos de magnesio II. En este modelo, uno de los iones de magnesio atrae un protón del carbono 3´, esto desestabiliza al grupo hidroxilo, permitiendo un ataque nuclefilico “el ataque de una molécula cargada negativamente total o parcialmente, de forma tal que atrae electrones de un electrófilo”. El nucleófilo en este caso es el oxígeno 3 del primer grupo fosfato del nucleótido que va a ser adicionado. El segundo magnesio estabiliza el grupo fosfato, promoviendo la liberación de dos de los tres grupos fosfato del nucleótido elegido por la polimeraza. La estabilización se refiere a una neutralización electrostática, los grupos magnesio al tener carga positiva son puentes que evitan que los oxígenos del desoxi-nucleótido se repelan entre sí.

Figura 8.14. Reiji Okazaki. (1930-1975) fue un biólogo molecular japonés conocido por sus investigaciones en la replicación del ADN y, especialmente, por describir el papel de los llamados fragmentos de Okazaki, que descubrió al trabajar con su esposa Tsuneko Okazaki en 1968.
Como consecuencia de la limitación químico-estructural de las polimerasas de ADN, una de las cadenas crece hacia la pinza de replicación, de forma tal que solo necesita un cebador para copiar. Sin embargo, para la otra hebra, dado que la enzima solo puede copiar en una dirección se crea un dilema bastante interesante. En esta hebra la polimeraza solo podría copias desde la pinza de replicación, lo que dejaría constantemente atrás al cebador, copiando un segmento y dejando al resto de la secuencia que se va abriendo paulatinamente sin copiar. ¿Cómo resolver este dilema estructural? La solución más sencilla sería la evolución de una enzima hipotética que lograra copiar desde la posición 5´, pero nuevamente esa proteína nunca se encontró. La respuesta a esta incógnita es el empleo de un mecanismo relativamente complicado de síntesis a pedazos o discontinua. Es por esto que la replicación del ADN se define como semidiscontinua, una hebra se replica continuamente y la otra discontinuamente.

Figura 8.15. Tsuneko Okazaki. (1933-viva a 2023) es una científica japonesa conocida por descubrir e investigar los fragmentos de Okazaki, junto con su marido Reiji Okazaki. Los fragmentos de Okazaki han contribuido a explicar la replicación del ADN. La doctora Okazaki ha continuado su relación con la academia, contribuyendo con más avances en la investigación del ADN. Graduada en la Universidad de Nagoya en 1956, Tsuneko Okazaki fue la primera mujer profesora en la universidad japonesa de Nagoya, y actualmente es profesora en el Instituto Médico de la Universidad Fujita. En el año 2000 recibió el Premio L'Oréal-UNESCO a Mujeres en Ciencia, que se concede anualmente a cinco científicas distinguidas, una por cada continente.
La hebra que puede sintetizarse desde un primer en dirección a la pinza de replicación es copiada de forma continua y es denominada Hebra Líder. La hebra que debe ser copiada a fragmentos se denomina Hebra Rezagada. Cabe destacar que esta nomenclatura no tiene nada que ver con la velocidad de replicación, estudios posteriores en cinética han demostrado que ambas cadenas se sintetizan más o menos a la misma velocidad, pero si es cierto que la cadena rezagada requiere de un mecanismo de replicación mucho más complejo que la cadena líder.
Ahora bien, ¿Qué hay sobre el problema de huevo-gallina entre los cebadores y las polimerasas? Recapitulando, in vitro las polimerasas de ADN son las proteínas que sintetizan el ADN, pero no pueden operar a menos que un pequeño fragmento de ADN una esté pegado a la hebra relajada. La polimerasa debe poder anclarse al extremo 3´libre del cebador para poder copiar. Lo cual nos trae a que, ¿cómo se sintetiza un cebador si las polimerasas de ADN lo necesitarían en primera instancia para poder formarlo? Los biólogos moleculares insinuaron que in vivo el proceso era diferente, y de hecho lo es. Estudios posteriores demostraron que los cebadores in vivo no están compuestos por ADN sino por ARN. El proceso involucra a una enzima llamada Primasa de ARN, la cual reconoce orígenes de replicación en la hebra y sintetiza el cebador de ARN. Luego la ADN polimerasa inicia a copiar desde la punta 3´de la secuencia de ARN.

Figura 8.16. Modelo de la replicación del ADN bacteriano (YouTube).
El descubrimiento de que una de las hebras se sintetizas a retazos fue realizada por el biólogo japonés Reiji Okazaki “1930-1975” y por su esposa Tsuneko Okazaki “1933-viva” de la Universidad de Nagoya en el año de 1968. Nuevamente, los experimentos involucraban marcar las cadenas crecientes de ADN con algún tipo de marcador, que después era medido por un instrumento apropiado. En este caso se empleó tritio en la timidina, un isótopo del hidrógeno radiactivo. Okazaki encontró que, si se incubaba por unos segundos una bacteria con timidina marcada con tritio y se las mataba inmediatamente, el ADN extraído podía verse como fragmentos cortos de unos 1000 a 2000 nucleótidos de largo. En contraste si las células se las dejaba crecer por un minute o dos, la mayoría de la timina marcada con tritio hacía parte de una cadena mucho más larga y continua. Estos resultados implican que, a gran velocidad, el ADN de la hebra rezagada se sintetizan en fragmentos que son ensamblados en una hebra única. Estos fragmentos han sido denominados para la posteridad como los Fragmentos de Okazaki. Lamentablemente el profesor Renji Okazaki murió a temprana edad debido a cáncer por la exposición a la radiación por la bomba detonada en su ciudad natal, Hiroshima.
¿Qué sucede con el cebador de ARN?, in vitro, los cebadores de ADN se convierten en parte de la nueva cadena copiada, sin embargo, in vivo el cebador de ARN debe ser reemplazado a riesgo de crear moléculas híbridas de ADN-ARN muy inestables y tendientes a colapsar. Para evitar esto se emplea una enzima llamada ADN polimerasa I, esta reemplaza los fragmentos de ARN por ADN. Finalmente, para ensamblar los fragmentos se emplea la ADN ligasa.
Resumen de la replicación de ADN
La replicación de ADN es un proceso semiconservativo en el cual las dos hebras de la molécula madre se relajan. Cuando una doble hélice de AADN se relaja esta se desenrosca y expone sus bases nitrogenadas de forma lineal. Aunque esto causa enroscamientos en el resto de la molécula, los nudos se resuelven mediante las topoisomerasas de ADN. Posteriormente cada hebra seguirá una ruta diferente. La hebra líder solo requiere de un cebador o primer para iniciar la síntesis, este cebador se sintetiza en un lugar de la secuencia llamado origen de replicación, el cual es reconocido por una ARN polimerasa, que sintetiza el primer. De esta forma la ADN polimerasa III se coloca en la punta 3´ del cebador de ARN y comienza a copiar de forma continua a lo largo de la hebra. La otra hebra debe ser copiada en fragmentos, por lo que la ARN polimerasas que sintetiza el primer debe reconocer de corma cíclica varios orígenes de replicación, la ADN polimerasa III reconoce estos fragmentos t los copia de vez en vez, o en fragmentos “llamados fragmentos de Okazaki”. Finalmente, los primer de ARN son reemplazados por una secuencia de ADN por medio de la enzima ADN ligasa.
9. La replicación del ADN 3, en eucariotas
9. La replicación del ADN 3, en eucariotas
La secuencia de nucleótidos, representados como un modelo de lenguaje de letras, de la secuencia del genoma humano llenarían un libro de aproximadamente un millón de páginas. Aunque tomó varios años para que cientos de investigadores secuenciaran inicialmente el genoma humano, una sola célula del núcleo, que mide aproximadamente 10 micrómetros de diámetro, puede copiar todo este ADN en unas pocas horas.
Modelos de análisis
Dado que las células eucariotas tienen genomas grandes y estructuras cromosómicas complejas, nuestra comprensión de la replicación en eucariotas ha quedado rezagada en comparación con la replicación en bacterias. Para resolver esto, se han desarrollado sistemas experimentales eucariotas que son similares a los utilizados durante décadas para estudiar la replicación bacteriana.
Estos sistemas incluyen el aislamiento de células mutantes de levadura y animales que no pueden producir productos génicos específicos necesarios para varios aspectos de la replicación, el análisis de la estructura y mecanismo de acción de las proteínas de replicación de especies de arcaicas y el desarrollo de sistemas in vitro donde la replicación puede ocurrir en extractos celulares o mezclas de proteínas purificadas.

Figura 9.1. Xenopus laevis es una especie de rana acuática originaria de África subsahariana. Son conocidas como ranas africanas de garras lisas debido a que sus dedos no tienen membranas interdigitales. Son grandes, alcanzando una longitud de hasta 20 cm, y su piel es de color marrón o verde grisáceo con manchas oscuras. Los machos tienen una cresta dorsal prominente durante la época de apareamiento. Son ampliamente utilizadas en la investigación científica debido a su capacidad para producir grandes huevos y su facilidad para criar en cautiverio. Xenopus laevis también es conocida por ser una especie invasora en algunas partes del mundo donde ha sido introducida accidental o intencionalmente.
Uno de los sistemas más valiosos ha utilizado Xenopus, una rana acuática que comienza la vida como un huevo enorme equipado con todas las proteínas necesarias para llevarlo a través de una docena de divisiones celulares muy rápidas. Se pueden preparar extractos de estos huevos de rana que replicarán cualquier ADN agregado, independientemente de la secuencia. Estos extractos de huevo de rana también pueden soportar la replicación y división mitótica de núcleos de mamíferos, lo que ha hecho que este sistema sin células sea particularmente útil. Se pueden usar anticuerpos para eliminar extractos de proteínas específicas y luego se puede probar la capacidad de replicación del extracto en ausencia de la proteína afectada.
Inicio de la replicación del ADN en una célula eucariota
En las células de E. coli, la replicación comienza en un solo sitio a lo largo del cromosoma circular único. Las células de organismos superiores pueden tener mil veces más ADN que esta bacteria, sin embargo, sus polimerasas incorporan nucleótidos en el ADN a tasas mucho más lentas.
Replicones
Para acomodar estas diferencias, las células eucariotas replican su genoma en pequeñas porciones, llamadas replicones.

Figura 9.2. Esta imagen de microscopía electrónica muestra el material genético de una célula de una mosca del género Drosophila. Se observa una pareja de horquillas de replicación (flechas) que se alejan entre sí en direcciones opuestas. Entre las dos horquillas se ven regiones de ADN recién replicado que ya están cubiertas por partículas nucleosómicas, que tienen una densidad aproximadamente igual a la de las hebras parentales que aún no han sido replicadas. En otras palabras, la imagen muestra cómo se están duplicando las moléculas de ADN en una célula en división, y cómo los nuevos fragmentos de ADN se van empaquetando con proteínas llamadas nucleosomas, para formar la estructura compacta que se conoce como cromatina.
Cada replicón tiene su propio origen desde el cual las horquillas de replicación proceden hacia afuera en ambas direcciones (ver Figura 9.2).
Número de replicones
En una célula humana, la replicación comienza en alrededor de 10 000 a 100 000 orígenes de replicación diferentes. La existencia de replicones fue demostrada por primera vez en experimentos autoradiográficos en los cuales se demostró que moléculas individuales de ADN se replican simultáneamente en varios sitios a lo largo de su longitud (ver Figura 13.19).

Figura 9.3. Este experimento demuestra que la replicación en los cromosomas eucariotas comienza en muchos sitios a lo largo del ADN. Las células se incubaron con una sustancia radioactiva llamada timidina tritiada ([3H] timidina) durante un breve periodo antes de preparar fibras de ADN para ser fotografiadas en una placa de autorradiografía. Las líneas de pequeños granos negros que se ven en la imagen indican los lugares donde el precursor de ADN radioactivo se incorporó durante el periodo de marcaje. Es evidente que la síntesis de ADN está ocurriendo en diferentes sitios separados a lo largo de la misma molécula de ADN. Como se muestra en el dibujo, la iniciación de la replicación comienza en el centro de cada sitio de incorporación de timidina, formando dos horquillas de replicación que se alejan entre sí hasta que se encuentran con una horquilla de replicación vecina.
Aproximadamente el 10 al 15 por ciento de los replicones están activamente comprometidos en la replicación en cualquier momento durante la fase S del ciclo celular. Los replicones ubicados cerca uno del otro en un cromosoma determinado se replican simultáneamente (Figura 9.3). Además, aquellos replicones activos en un momento particular durante una ronda de síntesis de ADN tienden a ser activos en un momento comparable en rondas sucesivas.
Las histonas
En las células de mamíferos, el momento de replicación de una región cromosómica se correlaciona aproximadamente con la actividad de los genes en la región y / o su estado de compactación. La presencia de histonas acetiladas, que está estrechamente correlacionada con la transcripción génica, es un factor probable en la determinación de la replicación temprana de los loci génicos activos.
Las histonas son proteínas que se encuentran en el núcleo de las células y que están involucradas en el empaquetamiento del ADN. Cuando se produce la acetilación de una histona, se agrega un grupo químico llamado acetilo a la proteína, lo que cambia su carga eléctrica y afecta su capacidad para interactuar con el ADN. En general, la acetilación de histonas está asociada con una mayor accesibilidad del ADN a los factores de transcripción y una mayor actividad génica.
Si utilizamos la analogía de una llave de encendido, podríamos decir que una histona acetilada está en "encendido". La acetilación de las histonas se asocia con una estructura cromatínica más relajada y accesible para la maquinaria de transcripción y replicación, lo que permite una mayor actividad génica y una replicación temprana en la célula. En cambio, la falta de acetilación (o desacetilación) de las histonas las "apaga", lo que resulta en una estructura cromatínica más compacta y un acceso reducido a la maquinaria de transcripción y replicación.
Las regiones del cromosoma más altamente compactadas y menos acetiladas se empaquetan en, y son las últimas regiones en replicarse. Esta diferencia en el momento de la replicación no está relacionada con la secuencia de ADN porque el cromosoma X inactivo y heterocromático en las células de los mamíferos femeninos se replica al final de la fase S, mientras que el cromosoma X activo y eucromático se replica en una etapa anterior. De manera similar, el locus de la globina β se replica temprano durante la fase S en las células eritroides donde el gen se expresa, pero mucho más tarde en las células no eritroides donde el gen permanece inactivo.
Formación de cromosomas
Aunque la desacetilación de las histonas es una de las modificaciones químicas que ocurren en las proteínas de la cromatina para formar los cromosomas, existen otros procesos para el correcto empaquetado de estas superestructuras. Por ejemplo, la fosforilación de las histonas también puede influir en la organización de la cromatina y la formación de los cromosomas. Además, la formación de un cromosoma implica la condensación de la cromatina y la organización de las proteínas de la estructura del cromosoma en una estructura altamente ordenada. Por lo tanto, la desacetilación de las histonas por sí sola no es suficiente para iniciar la formación de un cromosoma.

Figura 9.4. Saccharomyces cerevisiae es una especie de levadura unicelular que se utiliza ampliamente en la producción de alimentos y bebidas fermentadas como la cerveza, el vino y el pan. Es un organismo eucariota que tiene un tamaño de célula de aproximadamente 5 micrómetros y se reproduce por fisión binaria. La levadura de cerveza tiene un genoma de aproximadamente 12 millones de pares de bases y se han identificado más de 6,000 genes en su secuencia de ADN. Además de su uso industrial, S. cerevisiae es un organismo modelo muy importante en la investigación biológica y ha contribuido significativamente a la comprensión de procesos celulares como la replicación del ADN, la regulación génica y la respuesta al estrés.
Levaduras y vertebrados
Durante la última década, los científicos han estado estudiando cómo comienza la replicación en células eucariotas. Han hecho un gran progreso en este tema utilizando la levadura de cerveza S. cerevisiae, ya que pueden sacar los orígenes de replicación del cromosoma de la levadura y ponerlos en moléculas de ADN bacterianas, lo que les da la capacidad de replicar en una célula de levadura o en extractos celulares que contienen las proteínas de replicación necesarias. Estas secuencias se llaman secuencias de replicación autónomas (ARS). Si se muta la ARS de manera que no puede unirse al complejo de reconocimiento de origen (ORC), la iniciación de la replicación no puede ocurrir.
Ha sido más difícil estudiar los orígenes de replicación en células de vertebrados que en levaduras. Aunque se ha descubierto que el ADN de vertebrados no tiene secuencias específicas como las ARS, los estudios de la replicación de cromosomas intactos de mamíferos sugieren que la replicación comienza dentro de regiones definidas del ADN en lugar de seleccionarse aleatoriamente como en los extractos de huevos de anfibios. Se cree que una molécula de ADN contiene muchos sitios donde se puede iniciar la replicación, pero solo se usan algunos de estos sitios en una célula determinada en un momento dado.
Factores epigenéticos
Los factores epigenéticos locales, como la posición de los nucleosomas, los tipos de modificaciones de histonas, el estado de metilación del ADN, el grado de superenrollamiento y el nivel de transcripción, pueden influir en la selección de los sitios de iniciación de la replicación.
Limitando la replicación a una vez por ciclo celular
Es esencial que cada parte del genoma se replique una vez, y solo una vez, durante cada ciclo celular.
Consecuencias de no limitar la replicación
Si no se cumple la replicación una vez y solo una vez por ciclo celular, pueden ocurrir graves errores en la distribución del material genético a las células hijas, lo que puede llevar a problemas graves como mutaciones, aneuploidía (un número anormal de cromosomas) y disfunción celular. Estos errores pueden ser una causa importante de enfermedades genéticas y cáncer. Por lo tanto, es importante que el proceso de replicación del ADN sea cuidadosamente regulado y controlado para garantizar una distribución adecuada y precisa del material genético durante cada ciclo celular. Las consecuencias de una replicación inadecuada pueden ser graves y causar una variedad de enfermedades. Aquí hay una lista de algunas enfermedades comunes derivadas de errores en la replicación del ADN:
(a) Cáncer: El cáncer es causado por mutaciones en el ADN que hacen que las células se dividan sin control. Estas mutaciones pueden ser causadas por errores en la replicación del ADN que no se corrigen adecuadamente.
(b) Síndrome de Down: El síndrome de Down es una condición genética causada por la presencia de una copia extra del cromosoma 21. Esto puede ocurrir cuando se produce un error en la replicación del ADN durante la división celular.
(c) Enfermedades mitocondriales: Las enfermedades mitocondriales son causadas por mutaciones en el ADN mitocondrial, que se encuentra fuera del núcleo de la célula y se replica de manera diferente al ADN nuclear. Los errores en la replicación del ADN mitocondrial pueden causar enfermedades que afectan la función de los músculos y otros tejidos.
(d) Anemia de células falciformes: La anemia de células falciformes es una enfermedad genética que afecta la forma de los glóbulos rojos y puede causar dolor y otros problemas de salud. Esta enfermedad es causada por una mutación en el gen de la hemoglobina, que puede ser el resultado de un error en la replicación del ADN.
(e) Enfermedades autoinmunitarias: Las enfermedades autoinmunitarias ocurren cuando el sistema inmunológico del cuerpo ataca por error sus propios tejidos. Estas enfermedades pueden estar relacionadas con mutaciones genéticas que resultan de errores en la replicación del ADN.
Es importante tener en cuenta que muchas enfermedades tienen múltiples causas y que la replicación del ADN es solo una de las muchas cosas que pueden salir mal en el cuerpo. Sin embargo, la replicación adecuada del ADN es esencial para mantener la salud celular y prevenir una amplia gama de enfermedades.
Por lo tanto, debe existir algún mecanismo que impida la re-iniciación de la replicación en un sitio que ya ha sido duplicado. El inicio de la replicación en un origen particular requiere el paso del origen a través de varios estados distintos. Algunos de los pasos que ocurren en un origen de replicación en una célula de levadura se ilustran en la Figura 13.20.

Figura 9.5. Pasos necesarios para la replicación del ADN en una célula de levadura. Para iniciar la replicación, es esencial que cada origen de replicación del genoma de la célula se una una sola vez a la compleja proteína ORC (Complejo de Reconocimiento de Orígenes) durante el ciclo celular de la levadura. En el paso 2, seis proteínas diferentes (Mcm2-Mcm7) se unen al origen de replicación después de la mitosis y forman un complejo llamado pre-RC, que está "licenciado" para iniciar la replicación. Para que la replicación comience, se requiere la activación de una proteína quinasa llamada Cdk y una segunda quinasa llamada DDK en el paso 3.
Una vez iniciada la replicación, las proteínas Mcm2-Mcm7 se convierten en la helicasa replicativa que separa las dos hebras del ADN para que puedan ser copiadas. En el paso 4, la replicación ha avanzado un corto trayecto en ambas direcciones desde el origen. Los MCM complejos que se encuentran en la pre-RC forman una helicasa replicativa de ADN que separa las hebras en ambos extremos de la replicación.
En el paso 5, después de que ambas hebras originales del ADN se hayan replicado, el complejo ORC todavía está presente en ambos orígenes, y las proteínas de replicación, incluyendo las helicasas MCM, se han desplazado del ADN. En las células de levadura, las proteínas MCM se exportan del núcleo y la re-iniciación de la replicación no puede ocurrir hasta que la célula haya pasado por la mitosis.
En las células de vertebrados, hay varios mecanismos que impiden la re-iniciación de la replicación, como la liberación del complejo ORC después de su uso en la fase S, la actividad continua de Cdk desde la fase S hasta la mitosis, la fosforilación de Cdc6 y su posterior exportación del núcleo, y la degradación de Cdt1 o su inactivación por un inhibidor unido.
Pasos similares que requieren proteínas homólogas tienen lugar en plantas y animales, lo que indica que el mecanismo básico de inicio de la replicación se conserva entre los eucariotas.
Unión del origen de replicación a proteínas ORC
En el primer paso (Figura 9.5), el origen de replicación es unido por un complejo de proteínas ORC, que en las células de levadura permanece asociado con el origen durante todo el ciclo celular. El ORC se ha descrito como una "plataforma de aterrizaje molecular" debido a su papel en la unión de las proteínas necesarias en los pasos siguientes.
Ensamblado del complejo de prereplicación
El siguiente paso importante (paso 2) es el ensamblaje de un complejo proteína-ADN, llamado complejo pre-replicación (pre-RC), que está "licenciado" (competente) para iniciar la replicación. Los estudios sobre la formación del pre-RC se han centrado en un conjunto de seis proteínas relacionadas con MCM (Mcm2-Mcm7). Las proteínas MCM se cargan en el origen de replicación en una etapa tardía de la mitosis, o poco después, con la ayuda de factores accesorios que se han unido previamente al ORC.
Las seis proteínas Mcm2-Mcm7 interactúan entre sí para formar un complejo con forma de anillo hexamérico (de 6 miembros) (el complejo MCM) que posee actividad de helicasa (como en el paso 4, Figura 9.5). Existe una fuerte evidencia que sugiere que el complejo MCM es la helicasa replicativa eucariota, es decir, la helicasa responsable de desenrollar el ADN en la horquilla de replicación (análogo a DnaB en E. coli).
En la etapa pre-RC mostrada en el paso 2, cada uno de los orígenes contiene un complejo MCM hexamérico doble, es decir, dos helicasas replicativas completas, que permanecen inactivas en esta etapa del ciclo celular. Cada una de estas replicasas viajará en direcciones opuestas desde el origen una vez que comience la replicación.
Activación del complejo de pre-replicación
La formación de un pre-RC marca un lugar en el genoma como un posible origen de replicación, pero no garantiza que en realidad sea un lugar donde se iniciará la replicación. Durante la mayoría de los ciclos celulares, se ensamblan muchos más pre-RC de los que se van a utilizar y no está claro qué determina qué sitios potenciales de replicación se seleccionarán posteriormente. Independientemente del mecanismo de selección, justo antes del comienzo de la fase S del ciclo celular, la activación de proteínas quinasas clave conduce a la fosforilación del complejo MCM y otras proteínas y a la iniciación de la replicación en sitios seleccionados en el genoma (paso 3, Figura 9.5). Una de estas proteínas quinasas es una quinasa dependiente de ciclina (Cdk). La actividad de la Cdk permanece alta desde la fase S hasta la mitosis, lo que suprime la formación de nuevos complejos pre-replicación. Por lo tanto, cada origen solo puede activarse una vez por ciclo celular. La cesación de la actividad de la Cdk al final de la mitosis permite el ensamblaje de pre-RC para el próximo ciclo celular.
Una vez que comienza la replicación al comienzo de la fase S, la proteína MCM helicasa se mueve junto con la horquilla de replicación (paso 4), aunque no se sabe exactamente cómo funciona esta proteína con forma de anillo. Lo que sucede con las proteínas MCM después de la replicación depende de la especie estudiada. En células de mamíferos, las proteínas MCM se desplazan del ADN, pero aparentemente permanecen en el núcleo. Sin embargo, las proteínas MCM no pueden volver a asociarse con un origen de replicación que ya se ha activado.
Orquilla de replicación
La replicación del ADN es un proceso crucial en la vida de una célula, durante el cual se copia la información genética almacenada en el ADN. En las células eucariotas, la replicación del ADN ocurre en la fase S del ciclo celular y es un proceso complejo que involucra una serie de proteínas y enzimas.
Inicio
En las células eucariotas, la replicación del ADN comienza en sitios específicos llamados "orígenes de replicación", que están marcados por secuencias de ADN especiales. Una vez que se identifica un origen de replicación, se forma una estructura conocida como "orquilla de replicación", que es esencial para el proceso de replicación.
La orquilla de replicación eucariota es una estructura que se forma cuando se separan las dos hebras de ADN en un sitio específico de origen de replicación. A medida que la orquilla de replicación se mueve hacia abajo de la molécula de ADN, se producen dos horquillas de replicación en direcciones opuestas. Cada horquilla de replicación consiste en dos regiones distintas: la región de unión de ADN y la región de síntesis.

Figura 9.6. Orquilla o pinza de replicación eucariota.
Región de unión
En la región de unión de ADN, la hebra de ADN se separa en dos hebras individuales y se forma un complejo llamado replisoma. El replisoma está compuesto por una serie de proteínas que trabajan juntas para desenrollar y separar las dos hebras de ADN, y para mantener las hebras separadas mientras se produce la replicación. Las enzimas más relevantes involucradas en la replicación del ADN y que forman parte del replisoma son las siguientes:
(a) Helicasas: son enzimas que utilizan energía ATP para desenrollar las hebras de ADN y separarlas para permitir la replicación.
(b) Topoisomerasas: son enzimas que ayudan a relajar la tensión que se produce en el ADN cuando se está desenrollando y separando, para evitar que las hebras se rompan.
(c) Primasa: es una enzima que sintetiza un pequeño fragmento de ARN, conocido como cebador, que es necesario para que las polimerasas de ADN comiencen la síntesis de la nueva hebra de ADN.
(d) Polimerasas de ADN: son enzimas que sintetizan las nuevas hebras de ADN utilizando las hebras existentes como molde. En las células eucariotas, la polimerasa delta es la principal enzima responsable de la replicación del ADN, pero la polimerasa alfa, polimerasa epsilon y otras polimerasas de ADN también están involucradas.
(e) Ligasas: son enzimas que unen los fragmentos de ADN sintetizados por las polimerasas de ADN para formar la nueva hebra continua de ADN.
En conjunto, estas enzimas y proteínas trabajan en coordinación para asegurar que la replicación del ADN sea precisa y eficiente, y que se produzcan dos copias idénticas del material genético durante la división celular.
Región de síntesis
En la región de síntesis, las proteínas del replisoma trabajan junto con un complejo de enzimas llamado polimerasa de ADN para sintetizar una nueva hebra de ADN complementaria a cada hebra original. Las polimerasas de ADN son enzimas que catalizan la adición de nucleótidos a una hebra de ADN en crecimiento, y hay varios tipos diferentes de polimerasas de ADN involucrados en la replicación del ADN eucariota.
En particular, se sabe que cuatro tipos diferentes de polimerasas de ADN están involucrados en la replicación del ADN eucariota: polimerasa alfa, polimerasa beta, polimerasa gamma y polimerasa delta.
(a) La polimerasa alfa está involucrada en la síntesis de los cebadores de ARN necesarios para la replicación del ADN.
(b) La polimerasa beta está involucrada en la reparación del ADN.
(c) La polimerasa gamma está involucrada en la replicación del ADN mitocondrial.
(d) La polimerasa delta es la principal polimerasa involucrada en la replicación del ADN nuclear.
(e) La polimerasa epsilon es responsable de sintetizar la cadena líder de la nueva hebra de ADN en el replisoma eucariota, durante la replicación del ADN en la fase S del ciclo celular. La cadena líder es la cadena de la nueva hebra de ADN que se sintetiza de manera continua, mientras que la otra cadena, la cadena retrasada, se sintetiza de manera discontinua en fragmentos llamados Okazaki. Además, la polimerasa epsilon tiene la capacidad de corregir errores en la síntesis del ADN durante la replicación, a través de su actividad de exonucleasa. De esta manera, contribuye a asegurar la precisión y la fidelidad de la replicación del ADN.
En resumen, la orquilla de replicación eucariota es una estructura crucial para la replicación del ADN, y está compuesta por una serie de proteínas y enzimas que trabajan juntas para desenrollar, separar y replicar el ADN. La replicación del ADN en células eucariotas es un proceso complejo y altamente regulado, y las polimerasas de ADN juegan un papel importante en la síntesis de las hebras de ADN nuevas y complementarias
Comparación con la orquilla bacteriana
En comparación con la replicación del ADN eucariota, la replicación del ADN procariota es un proceso más simple que se lleva a cabo en un solo punto de origen de replicación y utiliza menos proteínas y enzimas.
En las células procariotas, la replicación del ADN comienza en un solo punto de origen de replicación y forma una estructura similar a una burbuja de replicación que se expande en ambas direcciones a medida que se sintetiza el ADN. La orquilla de replicación procariota es menos compleja que la eucariota y utiliza menos proteínas y enzimas. En lugar de tener múltiples polimerasas de ADN, las células procariotas utilizan una sola polimerasa de ADN que es capaz de sintetizar ambas hebras del ADN en la dirección 5' a 3'.
Una de las principales diferencias entre la replicación del ADN eucariota y procariota es el uso de múltiples polimerasas de ADN en la replicación eucariota. En las células eucariotas, la polimerasa delta es la principal enzima responsable de la replicación del ADN, pero la polimerasa alfa, polimerasa epsilon y otras polimerasas de ADN también están involucradas en la replicación. Cada una de estas polimerasas de ADN tiene diferentes propiedades y funciones, lo que les permite trabajar juntas de manera eficiente para sintetizar nuevas hebras de ADN.
En contraste, en las células procariotas, una sola polimerasa de ADN es responsable de la replicación del ADN. Esta polimerasa es capaz de sintetizar ambas hebras del ADN en la dirección 5' a 3'. Además, la replicación del ADN en células procariotas es más rápida que en células eucariotas debido a la menor complejidad de la maquinaria de replicación.
En conclusión, la orquilla de replicación es una estructura fundamental para la replicación del ADN en células eucariotas. A través de la acción de proteínas y enzimas, la orquilla de replicación es capaz de separar y replicar las hebras de ADN de manera eficiente y precisa. En comparación con la replicación del ADN en células procariotas, la replicación del ADN eucariota es más compleja y utiliza múltiples polimerasas de ADN para sintetizar nuevas hebras de ADN.
La replicación del ADN y la estructura nuclear
Cuando el ADN se replica en una célula, no ocurre de manera aleatoria en el núcleo, sino que se concentra en 50 a 250 lugares específicos llamados focos de replicación (Figura 9.7). En cada uno de estos focos, hay muchos puntos de replicación, que son como pequeñas máquinas que trabajan juntas para crear nuevas hebras de ADN. Se estima que cada una de las regiones rojas brillantes en la Figura 9.7 contiene alrededor de 10 a 100 puntos de replicación trabajando al mismo tiempo.

Figura 9.7. Demostración de que la replicación del ADN no ocurre al azar en todo el núcleo, sino que se concentra en lugares específicos. Antes de que comience la síntesis del ADN en la fase S, se juntan diversos factores necesarios para comenzar la replicación en lugares concretos dentro del núcleo, formando lo que se conoce como centros de pre-replicación. Estos lugares se ven como objetos rojos y brillantes en la imagen microscópica, que ha sido teñida con un anticuerpo fluorescente contra el factor de replicación A (RPA), una proteína que se une al ADN de una sola hebra y es necesaria para la replicación. Otros factores importantes para la replicación, como la PCNA y la compleja enzima polimerasa-primasa, también se concentran en estos focos. En otras palabras, el ADN no se replica de manera desordenada en todo el núcleo de la célula, sino que hay lugares específicos en los que se concentran las proteínas necesarias para comenzar la replicación. Estos lugares se ven como puntos brillantes en una imagen microscópica y se llaman centros de pre-replicación. El hecho de que las proteínas se concentren en estos centros ayuda a asegurar que la replicación del ADN sea precisa y completa en cada célula.
La agrupación de los puntos de replicación puede servir para coordinar la replicación de las partes adyacentes del cromosoma. Esto ayuda a garantizar que el ADN se replique de manera precisa y completa en cada célula, para que las células hijas tengan la información genética necesaria para funcionar correctamente. En resumen, la célula utiliza una estructura organizada para replicar el ADN y garantizar que la información genética se transmita de manera confiable a las células hijas.
Cambios en la estructura de la cromatina
Durante la replicación del ADN en células eucariotas, la cromatina experimenta una serie de cambios importantes que son esenciales para el éxito de la replicación.
Despliegue
En primer lugar, la cromatina se desenrolla para permitir el acceso de la maquinaria de replicación al ADN subyacente. Este proceso es facilitado por las histonas, que se desplazan hacia los extremos de los nucleosomas para permitir la exposición del ADN. La descompactación de la cromatina también se ve facilitada por enzimas que catalizan la rotura temporal de los enlaces entre las histonas y el ADN.
Inicio de replicación
Una vez que la cromatina ha sido desenrollada y expuesta, se procede a la iniciación de la replicación. Esto implica la formación de una burbuja de replicación, donde se separan temporalmente las dos hebras del ADN. A medida que la maquinaria de replicación se mueve a lo largo de la molécula de ADN, la burbuja de replicación se expande para permitir la continuación de la replicación. La formación de la burbuja de replicación está controlada por una serie de proteínas que interactúan con las secuencias de origen de replicación del ADN.
Replicación
Una vez que se ha iniciado la replicación, las enzimas de replicación, como la ADN polimerasa, trabajan para sintetizar nuevas hebras de ADN complementarias a las hebras originales. Estas enzimas se unen a la hebra de ADN de origen y avanzan a lo largo de ella, agregando nucleótidos de forma secuencial y construyendo una nueva cadena de ADN complementaria. En la hebra líder, la polimerasa se mueve continuamente a lo largo del ADN mientras que en la hebra rezagada se forma un fragmento de Okazaki.
Repliegue
Finalmente, una vez que se ha completado la replicación del ADN, la cromatina debe volver a su estado compactado normal para que se puedan llevar a cabo las funciones celulares normales. Esto implica la reintegración de las histonas en la cromatina y la compactación del ADN en nucleosomas y fibras de cromatina más grandes.
Errores
Si estos procesos no se llevan a cabo adecuadamente, pueden ocurrir una serie de problemas en la célula. Por ejemplo, si la cromatina no se desenrolla adecuadamente, la maquinaria de replicación no podrá acceder al ADN subyacente y la replicación no se llevará a cabo. Del mismo modo, si se producen errores en la replicación del ADN, como la inserción incorrecta de nucleótidos, esto puede dar lugar a mutaciones genéticas y enfermedades. En general, la replicación del ADN y los cambios asociados en la cromatina son procesos críticos para la supervivencia y el buen funcionamiento de la célula.
Resumen
La replicación del ADN en las células eucariotas es un proceso complejo y altamente regulado que comienza con la iniciación de la replicación en sitios específicos del genoma. Durante la replicación, la ADN polimerasa sintetiza nuevas hebras de ADN a partir de una plantilla de ADN existente. Las hebras de ADN se separan en la horquilla de replicación, y cada hebra sirve como molde para la síntesis de una nueva hebra complementaria.
La replicación de la cadena líder se realiza de manera continua, mientras que la cadena rezagada se replica en fragmentos de Okazaki que luego se unen para formar una hebra completa. Una serie de enzimas y proteínas trabajan juntas en la maquinaria de replicación, incluyendo las polimerasas alfa, delta y epsilon, la helicasa, la primasa, la topoisomerasa y la ligasa.
Además, durante la replicación del ADN, se producen cambios en la estructura de la cromatina para permitir el acceso de la maquinaria de replicación a las regiones de ADN. Si se produce un error durante la replicación del ADN, pueden activarse mecanismos de reparación para corregir los errores. La replicación del ADN es fundamental para la transmisión precisa de la información genética de una célula madre a sus células hijas y es esencial para la supervivencia y la función normal de los organismos eucariotas.
10. Reparación del ADN
10. Reparación del ADN
La vida en la Tierra está constantemente expuesta a fuerzas destructivas que pueden dañar los organismos.
Factores que afectan la estabilidad del ADN
El ADN, que es la molécula que contiene la información genética, es una de las moléculas más vulnerables a estos daños. Por un lado, es muy importante que la información genética no cambie mucho al ser transmitida de célula en célula o de individuo en individuo. Pero, por otro lado, el ADN es susceptible a daños producidos por el ambiente, como:
(a) Radiación ionizante: Este tipo de radiación puede romper los enlaces en la cadena de ADN, lo que resulta en daños en el material genético. La exposición prolongada a la radiación ionizante puede aumentar el riesgo de desarrollar enfermedades como el cáncer. Es importante tener en cuenta que la radiación ionizante no solo proviene del ambiente (como los rayos cósmicos), sino que también puede ser producida artificialmente, por ejemplo, en procedimientos médicos como la radioterapia.
(b) Químicos reactivos externos: Muchas sustancias químicas, tanto naturales como sintéticas, pueden reaccionar con el ADN y causar daños. Algunos de estos químicos son producidos por la propia célula durante el metabolismo normal, mientras que otros pueden provenir del ambiente, como los productos químicos tóxicos en el aire y el agua. Estos químicos pueden causar alteraciones en la estructura de la molécula de ADN y/o provocar mutaciones.
(c) Radiación ultravioleta: La exposición al sol es una fuente importante de radiación ultravioleta (UV), que puede ser muy dañina para el ADN. Los rayos UV pueden causar daños en las bases nitrogenadas de la molécula de ADN, lo que puede provocar mutaciones. Las mutaciones en el ADN pueden conducir a la formación de células cancerosas, y también pueden ser responsables del envejecimiento prematuro de la piel.
(d) Energía generada por el metabolismo: La energía producida por el metabolismo celular también puede dañar el ADN. En condiciones normales, los procesos de reparación del ADN son suficientes para corregir estos daños. Sin embargo, si estos daños no se reparan adecuadamente, pueden acumularse con el tiempo y aumentar el riesgo de mutaciones y cáncer.
(e) Metabolitos secundarios: algunos organismos producen metabolitos secundarios que pueden dañar el ADN. Por ejemplo, las aflatoxinas producidas por ciertos tipos de hongos pueden causar daño al ADN y aumentar el riesgo de cáncer hepático.

Figura 10.1. Existen varias relaciones entre el estilo de vida, la reparación del ADN y el cáncer. Por ejemplo, ciertos hábitos de vida, como el tabaquismo y la exposición prolongada a la radiación ultravioleta, pueden aumentar el riesgo de desarrollar cáncer al dañar el ADN. Sin embargo, los sistemas de reparación del ADN en nuestro cuerpo generalmente pueden detectar y reparar estos daños para evitar la aparición de células cancerosas.
Por otro lado, las mutaciones que ocurren en los genes responsables de la reparación del ADN pueden aumentar el riesgo de cáncer. Si estas mutaciones se heredan o se adquieren durante la vida, pueden conducir a una disminución de la capacidad del cuerpo para reparar el ADN dañado, lo que a su vez aumenta la probabilidad de que se desarrollen células cancerosas.
Además, la eficacia de los tratamientos contra el cáncer, como la quimioterapia y la radioterapia, depende en gran medida de la capacidad de las células cancerosas para reparar el ADN dañado. Si las células cancerosas tienen mutaciones que afectan su capacidad para reparar el ADN, estos tratamientos pueden ser más efectivos para matarlas.
En resumen, la relación entre el estilo de vida, la reparación del ADN y el cáncer es compleja y multifacética. La investigación continua en este campo es fundamental para comprender mejor cómo se pueden prevenir y tratar los cánceres relacionados con el ADN y mejorar los tratamientos existentes.
(f) Iones superóxido: los iones superóxido son especies reactivas de oxígeno que se producen como resultado del metabolismo celular. Estos iones pueden causar daño al ADN y otros componentes celulares al reaccionar con ellos de manera no específica.
En resumen, aunque el ADN es una molécula esencial para la vida, también es vulnerable a muchos tipos de daños causados por factores ambientales y celulares. Es por eso que las células tienen mecanismos para reparar estos daños y mantener la integridad del ADN. Sin estos mecanismos, el ADN podría sufrir cambios permanentes que podrían tener graves consecuencias para la salud y la supervivencia de la célula y el organismo.
La mutación
Estos daños son conocidos como lesiones y pueden provocar cambios permanentes en el ADN, llamados mutaciones. Las mutaciones pueden tener consecuencias graves, como interferir con la transcripción y la replicación del ADN, causar la transformación maligna de una célula o acelerar el proceso de envejecimiento de un organismo. Por lo tanto, es esencial que las células tengan mecanismos para reparar el daño del ADN.
Afortunadamente, las células tienen una amplia variedad de sistemas de reparación que pueden corregir prácticamente cualquier tipo de daño al que el ADN pueda estar expuesto.
De hecho, es muy raro que una mutación pase desapercibida por estos sistemas de reparación. La existencia de estos sistemas demuestra cómo los mecanismos moleculares mantienen la homeostasis celular. La importancia de la reparación del ADN se puede apreciar en los efectos que tienen las deficiencias en los sistemas de reparación del ADN en los seres humanos.
Frecuencia de mutación en humanos
Es cierto que las células poseen sistemas de reparación altamente eficientes, pero no son infalibles. Aún así, la tasa de mutaciones en una población humana sana es relativamente baja, estimada en aproximadamente 70 mutaciones por generación. La mayoría de estas mutaciones son neutras y no tienen ningún efecto discernible en la salud o el desarrollo del individuo.
Mutaciones semiletales
Sin embargo, algunas mutaciones pueden ser perjudiciales para la salud, como las mutaciones semiletáles que reducen la viabilidad o la fertilidad de un organismo. Un ejemplo de esto es la fibrosis quística, una enfermedad genética que afecta la función pulmonar y digestiva y que es causada por una mutación en el gen CFTR.
Mutaciones letales
Otras mutaciones son letales y causan la muerte del individuo, como la anemia de células falciformes, una enfermedad genética en la que las células sanguíneas deformadas se obstruyen en los vasos sanguíneos, lo que causa dolor, daño a los órganos y puede ser fatal.
Mutaciones benéficas
Por último, aunque son menos comunes, también existen mutaciones beneficiosas que pueden proporcionar una ventaja selectiva en determinados entornos. Por ejemplo, la mutación en el gen que produce la hemoglobina en la población africana que vive en zonas con malaria, que les brinda cierta protección contra la enfermedad.

Figura 10.2. Los ojos azules son una característica relativamente rara en los seres humanos y se cree que su aparición se debe a una mutación en el gen OCA2, que se encuentra en el cromosoma 15. Esta mutación reduce la producción de melanina en el iris del ojo, lo que da como resultado el color azul. La frecuencia de esta mutación varía según la población. Por ejemplo, en Europa del Norte, aproximadamente el 80% de la población tiene ojos azules o verdes, mientras que en el sur de Europa, esta cifra es mucho menor, alrededor del 20%. No está claro si la selección sexual ha desempeñado un papel en la aparición de los ojos azules. El modelo del fugitivo loco de Fisher sugiere que ciertas características se vuelven atractivas para el sexo opuesto simplemente porque son raras o inusuales, lo que las convierte en un signo de buena salud y capacidad de supervivencia. Sin embargo, la evidencia de que la selección sexual haya influido en la aparición de los ojos azules en el ser humano es limitada.
En resumen, aunque los sistemas de reparación del ADN son muy eficientes, las mutaciones aún ocurren en una frecuencia relativamente baja en una población humana sana. La mayoría de estas mutaciones son neutras, aunque algunas pueden ser perjudiciales o beneficiosas para la salud del individuo.
Mecanismos de reparación del ADN bacteriano
El ADN es una molécula fundamental en la vida de los seres vivos, ya que contiene la información genética necesaria para el correcto funcionamiento y desarrollo de los organismos. Sin embargo, el ADN está expuesto a numerosas amenazas, como la radiación, los productos químicos y los errores durante la replicación, que pueden causar daños en su estructura y en su secuencia. Estos daños pueden resultar en mutaciones genéticas y, en algunos casos, pueden ser fatales para la célula o el organismo.
Las bacterias, siendo organismos unicelulares, están expuestas a ambientes hostiles y cambiantes, por lo que han desarrollado una serie de mecanismos de reparación del ADN para hacer frente a los daños. En este artículo, se describirán los principales mecanismos de reparación del ADN bacteriano.
(a) Uno de los mecanismos de reparación más comunes es el sistema de reparación por escisión de bases (BER, por sus siglas en inglés). Este sistema repara daños en una sola base, como la eliminación de bases oxidadas o alquiladas. El proceso comienza cuando una enzima, una glicosilasa, detecta el daño en una base y la elimina, creando un sitio de AP (apurínico o apirimidínico) en la cadena de ADN. Luego, otras enzimas se unen al sitio AP para reemplazar la base faltante y sellar la brecha en la cadena de ADN.
(b) Otro sistema de reparación es el sistema de reparación por escisión de nucleótidos (NER, por sus siglas en inglés), que repara daños en la estructura del ADN causados por la exposición a la radiación UV, productos químicos y otros factores ambientales. El proceso comienza cuando un complejo proteico reconoce y se une al daño en el ADN. Luego, una serie de enzimas cortan el ADN dañado, eliminando un segmento corto de ADN que contiene el daño. Finalmente, las enzimas de reparación reemplazan el segmento de ADN dañado por uno nuevo y lo unen a la cadena de ADN existente.
(c) Además de estos sistemas de reparación, las bacterias también tienen un sistema de reparación por recombinación homóloga (HR, por sus siglas en inglés), que repara roturas de doble cadena en el ADN causadas por la radiación y otros factores ambientales. En este proceso, una molécula de ADN sin daño, llamada molécula donante, se utiliza como plantilla para reparar el ADN dañado. La molécula donante se une a la cadena de ADN dañada y actúa como una plantilla para reconstruir el ADN dañado.
En resumen, las bacterias tienen una variedad de sistemas de reparación del ADN que les permiten hacer frente a los daños causados por factores ambientales. Estos mecanismos de reparación son esenciales para mantener la integridad genética y la supervivencia de las células bacterianas.
Mecanismos de reparación del ADN arcaico
En el caso de los organismos arqueanos, que pertenecen al dominio Archaea, existen varios mecanismos de reparación del ADN que les permiten enfrentar y sobrevivir en condiciones extremas. Los arqueanos habitan en ambientes hostiles como fuentes termales, aguas salinas y sedimentos, donde están expuestos a altas temperaturas, alta radiación y productos químicos tóxicos.
(a) Uno de los principales mecanismos de reparación del ADN en los arqueanos es la reparación por escisión de nucleótidos (NER, por sus siglas en inglés). Este mecanismo se encarga de reparar los daños causados por la radiación ultravioleta, los productos químicos tóxicos y otros agentes que pueden dañar el ADN. La NER implica la eliminación de un segmento corto de ADN que contiene el daño y su sustitución por un fragmento de ADN nuevo y sano.
(b) Otro mecanismo de reparación del ADN en los arqueanos es la reparación por escisión de bases (BER, por sus siglas en inglés). Este mecanismo se encarga de reparar los daños causados por productos químicos oxidantes y otras sustancias que pueden alterar las bases del ADN. La BER implica la eliminación de la base dañada y su sustitución por una nueva.
(c) Además de la NER y la BER, los arqueanos también cuentan con mecanismos de reparación del ADN como la reparación por recombinación homóloga (HR, por sus siglas en inglés) y la reparación por recombinación no homóloga (NHEJ, por sus siglas en inglés). La HR se encarga de reparar los daños causados por roturas en el ADN, y consiste en la utilización de una molécula de ADN complementaria para reparar la rotura. La NHEJ, por otro lado, se encarga de reparar las roturas del ADN sin la necesidad de una molécula complementaria, y es un mecanismo más utilizado por los organismos eucariotas que por los arqueanos.
En resumen, los organismos arqueanos cuentan con una amplia variedad de mecanismos de reparación del ADN que les permiten enfrentar y sobrevivir en ambientes hostiles. La NER y la BER son dos de los principales mecanismos de reparación utilizados por los arqueanos, aunque también utilizan la HR y la NHEJ para reparar los daños del ADN. Gracias a estos mecanismos de reparación del ADN, los arqueanos pueden mantener la integridad del material genético y sobrevivir en condiciones extremas.
Mecanismos de reparación del ADN eucariota
En las células eucariotas, existen varios mecanismos de reparación del ADN que se dividen en dos categorías principales: reparación por escisión y reparación por recombinación.
(a) La reparación por escisión se refiere a la eliminación de una sección del ADN dañado y su reemplazo con una nueva secuencia de ADN. Hay tres tipos principales de reparación por escisión: reparación por escisión de nucleótidos (NER), reparación por escisión de bases (BER) y reparación por escisión de desajuste (MMR).
(b) La reparación por escisión de nucleótidos es un proceso que implica la eliminación de un fragmento de ADN dañado y su reemplazo por una nueva secuencia de ADN. Este proceso es importante para la reparación de lesiones causadas por la radiación ultravioleta y la radiación ionizante. También es esencial para la reparación de lesiones químicas que causan deformaciones en la estructura del ADN, como los aductos de ADN.
(c) La reparación por escisión de bases es un proceso que elimina las bases dañadas del ADN y las reemplaza por nuevas bases. Este proceso es importante para la reparación de lesiones químicas que alteran las bases del ADN, como la oxidación o la alquilación.
(d) La reparación por escisión de desajuste se refiere a la eliminación y reemplazo de errores de apareamiento de bases durante la replicación del ADN. Este proceso es esencial para prevenir mutaciones y mantener la integridad del genoma.
(e) La reparación por recombinación implica la reparación del ADN a través del uso de una secuencia de ADN homóloga. Este proceso es importante para la reparación de roturas de doble cadena del ADN, que son causadas por factores como la radiación ionizante y los productos químicos. La recombinación homóloga también es esencial para la corrección de errores durante la replicación del ADN y para la reparación de las secuencias del ADN que se han perdido o dañado.
(f) Además de los mecanismos de reparación del ADN mencionados anteriormente, las células eucariotas también tienen sistemas de vigilancia que monitorean constantemente el genoma en busca de daños. Si se detecta un daño, se activan las vías de señalización que conducen a la reparación del ADN o a la muerte celular programada si el daño no puede ser reparado.
Importancia de la investigación en los mecanismos eucariotas
En conclusión, los mecanismos de reparación del ADN en las células eucariotas son esenciales para mantener la integridad del genoma y prevenir mutaciones que pueden conducir a enfermedades y trastornos. La investigación continua en estos mecanismos es fundamental para comprender mejor cómo se mantienen la investigación continua en estos mecanismos es fundamental para comprender mejor cómo se mantienen la integridad y la estabilidad genética en las células eucariotas. Además, esta investigación es crucial para desarrollar terapias efectivas para enfermedades relacionadas con daños en el ADN, como el cáncer.
Algunas terapias ya están en uso clínico, como los inhibidores de PARP para tratar el cáncer de mama y de ovario. Estos fármacos aprovechan la dependencia de las células tumorales de los mecanismos de reparación del ADN mediante la inhibición de la PARP, una enzima clave en la reparación de roturas de cadena simple de ADN. Sin embargo, se necesitan más investigaciones para desarrollar terapias más efectivas y menos tóxicas.
También es importante destacar que la reparación del ADN no solo es importante para la salud humana, sino también para la agricultura y la biotecnología. La ingeniería genética se ha convertido en una herramienta poderosa para modificar los organismos y crear nuevas características beneficiosas, pero también puede causar daños en el ADN. Por lo tanto, la comprensión de los mecanismos de reparación del ADN es crucial para la creación de organismos genéticamente modificados más seguros y efectivos.
En resumen, los mecanismos de reparación del ADN eucariota son complejos y esenciales para mantener la integridad y la estabilidad genética. Los avances en nuestra comprensión de estos mecanismos no solo nos ayudarán a entender mejor la biología celular, sino que también nos permitirán desarrollar terapias más efectivas y crear organismos genéticamente modificados más seguros y beneficiosos.
Relaciones evolutivas en los mecanismos de reparación del ADN
Es importante señalar que, aunque hay algunas similitudes en los mecanismos de reparación del ADN en todos los organismos, también hay muchas diferencias. Estas diferencias están relacionadas con las adaptaciones específicas de cada grupo de organismos a su entorno. Por ejemplo, las bacterias que habitan en entornos ricos en radiación han desarrollado sistemas de reparación de ADN que son más eficaces contra los daños causados por la radiación. Del mismo modo, las arqueas que habitan en entornos extremos, como los géiseres de Yellowstone, han desarrollado mecanismos de reparación que les permiten sobrevivir a las condiciones extremas en las que viven.
En términos evolutivos, se cree que los mecanismos de reparación de ADN más simples son los que se encuentran en las bacterias. Estos sistemas están presentes en organismos unicelulares y no tienen núcleos celulares. A medida que la complejidad de los organismos aumentó, también lo hizo la complejidad de los mecanismos de reparación de ADN. Las arqueas, que son organismos unicelulares pero más complejos que las bacterias, tienen sistemas de reparación de ADN más avanzados que los de las bacterias. Por último, los eucariotas, que son organismos multicelulares con núcleos celulares, tienen los sistemas de reparación de ADN más complejos.
En términos de relación entre bacterias y arqueas, se cree que las arqueas son más cercanas a los eucariotas que a las bacterias en términos de la evolución de los mecanismos de reparación del ADN. De hecho, se cree que algunos de los sistemas de reparación de ADN de las arqueas son más parecidos a los de los eucariotas que a los de las bacterias.
En conclusión, los mecanismos de reparación del ADN son una función esencial en todos los organismos vivos. A medida que la complejidad de los organismos aumenta, también lo hace la complejidad de los mecanismos de reparación de ADN. Sin embargo, hay muchas diferencias en los mecanismos de reparación de ADN entre diferentes grupos de organismos, y estas diferencias están relacionadas con las adaptaciones específicas de cada grupo a su entorno. La investigación continua en estos mecanismos es fundamental para comprender mejor cómo se mantienen la integridad del ADN y la homeostasis celular en diferentes organismos vivos.
11. Introducción a la síntesis de proteínas
11. Introducción a la síntesis de proteínas
En muchos sentidos, el progreso en la biología durante el siglo pasado se refleja en nuestro concepto cambiante del gen. Como resultado del trabajo de Mendel, los biólogos aprendieron que los genes son elementos discretos que rigen la aparición de rasgos específicos. Los mendelianos defendieron el concepto de que un gen determina un rasgo. Boveri, Weismann, Sutton y sus contemporáneos descubrieron que los genes tienen una encarnación física como partes del cromosoma. Morgan, Sturtevant y sus colegas demostraron que los genes tienen ubicaciones específicas: residen en ubicaciones particulares en cromosomas particulares, y estas ubicaciones permanecen constantes de un individuo de una especie a la siguiente. Griffith, Avery, Hershey y Chase demostraron que los genes estaban compuestos de ADN, y Watson y Crick resolvieron el rompecabezas de la estructura del ADN, que explicaba cómo esta notable macromolécula podría codificar información hereditaria. Aunque las formulaciones de estos conceptos fueron hitos en el camino hacia la comprensión genética, ninguno de ellos abordó el mecanismo por el cual la información almacenada en un gen se pone a funcionar para gobernar las actividades celulares. Este es el tema principal que se discutirá en el presente capítulo. Comenzaremos con conocimientos adicionales sobre la naturaleza de un gen, lo que nos acerca a su papel en la expresión de rasgos heredados.

Figura 11.1. De izquierda a derecha, primera fila: Archibald Garrod, George Beadle; segunda fila: Edward Tatum y François Jacob.
La relación ADN y proteínas
La primera percepción significativa sobre la función genética fue obtenida por Archibald Garrod 1857-1936, un médico escocés que informó en 1908 que los síntomas exhibidos por personas con ciertas enfermedades hereditarias raras fueron causados por la ausencia de enzimas específicas. Una de las enfermedades investigadas por Garrod fue la alcaptonuria, una afección que se diagnostica fácilmente porque la orina se oscurece al exponerse al aire. Garrod descubrió que las personas con alcaptonuria carecían de una enzima en su sangre que oxidaba el ácido homogentísico, un compuesto formado durante la descomposición de los aminoácidos fenilalanina y tirosina. A medida que se acumula el ácido homogentísico, se excreta en la orina y se oscurece en color cuando se oxida por el aire. Garrod había descubierto la relación entre un defecto genético, una enzima específica y una condición metabólica específica. Llamó a tales enfermedades "errores innatos del metabolismo". Como parece haber sucedido con otras observaciones tempranas de importancia básica en genética, los hallazgos de Garrod no fueron apreciados durante décadas.
La idea de que los genes dirigen la producción de enzimas fue resucitada en la década de 1940 por George Beadle 1903-1989 y Edward Tatum 1909-1975 del Instituto de Tecnología de California. Estudiaron Neurospora, un moho de pan tropical que crece en un medio muy simple que contiene una sola fuente de carbono orgánico (por ejemplo, un azúcar), sales inorgánicas y biotina (vitamina B). Debido a que necesita tan poco para vivir, se supuso que Neurospora sintetiza todos sus metabolitos. Beadle y Tatum razonaron que un organismo con una capacidad sintética tan amplia debería ser muy sensible a las deficiencias enzimáticas, que deberían detectarse fácilmente utilizando el protocolo experimental adecuado.
El plan de Beadle y Tatum era irradiar esporas de moho y detectar mutaciones que causaran que las células carecieran de una enzima particular. Para detectar estas mutaciones, se probaron las esporas irradiadas para determinar su capacidad de crecer en un medio mínimo que carecía de los compuestos esenciales conocidos por ser sintetizados por este organismo. Si una espora no puede crecer en un medio mínimo pero una espora genéticamente idéntica puede crecer en un medio suplementado con una coenzima particular (p. Ej., Ácido pantoténico de la coenzima A), los investigadores podrían concluir que las células tienen una deficiencia enzimática que les impide de sintetizar este compuesto esencial.
Un gen, una proteína y la definición del gen
Beadle y Tatum comenzaron irradiando más de mil células. Dos de las esporas demostraron ser incapaces de crecer en el medio mínimo: una necesitaba piridoxina (vitamina B6) y la otra necesitaba tiamina (vitamina B1). Eventualmente, se probó la progenie de aproximadamente 100 000 esporas irradiadas, y se aislaron docenas de mutantes. Cada mutante tenía un defecto genético que producía una deficiencia enzimática que impedía que las células catalizaran una reacción metabólica particular. Los resultados fueron claros: un gen lleva la información para la construcción de una enzima particular. Esta conclusión se conoció como la hipótesis de "un gen-una enzima". Una vez que se supo que las enzimas a menudo están compuestas de más de un polipéptido, cada uno de los cuales está codificado por su propio gen, el concepto se modificó a "un polipéptido un gen". Aunque esta relación sigue siendo una aproximación cercana de la función básica de un gen, también ha tenido que ser modificado debido al descubrimiento de que un solo gen a menudo genera una variedad de polipéptidos, principalmente como resultado de un empalme “splicing” alternativo. También sería evidente que la descripción de un gen estrictamente como un almacén de información para polipéptidos es una definición demasiado estricta. Muchos genes codifican moléculas de ARN que, en lugar de contener información para la síntesis de polipéptidos, funcionan como ARN en sí mismas. Con esto en mente, podría ser mejor definir un gen como un segmento de ADN que contiene la información para una cadena polipeptídica única o para uno o más ARN funcionales.

Figura 11.2. De izquierda a derecha: Jacques Monod, Sydney Brenner y Matthew Meselson.
Flujo de la información genética
La información presente en un segmento de ADN se pone a disposición de la célula mediante la formación de una molécula de ARN. La síntesis de un ARN a partir de una plantilla de ADN se denomina transcripción. El término transcripción denota un proceso en el que la información codificada en las cuatro letras de desoxirribonucleótidos de ADN se reescribe, o se transcribe, en un lenguaje similar compuesto por cuatro letras de ribonucleósidos de ARN. Examinaremos el mecanismo de la transcripción en breve, pero primero continuaremos con el papel de un gen en la formación de polipéptidos.
Los estudios llevados a cabo en la década de 1950 descubrieron la relación entre la información genética y la secuencia de aminoácidos, pero este conocimiento en sí mismo no proporcionaba ninguna pista sobre el mecanismo por el cual se genera la cadena polipeptídica específica. Como ahora sabemos, hay un intermedio entre un gen y su polipéptido; el intermediario es el ARN mensajero (ARNm). El descubrimiento trascendental del ARNm fue realizado en 1961 por François Jacob 1920-2013 y Jacques Monod 1910-1976 del Instituto Pasteur en París, Sydney Brenner 1927-Vivo de la Universidad de Cambridge y Matthew Meselson 1930-vivo del Instituto de Tecnología de California. Un ARN mensajero se ensambla como una copia complementaria de una de las dos cadenas de ADN que componen un gen. Debido a que su secuencia de nucleótidos es complementaria a la del gen del cual se transcribe, el ARNm conserva la misma información para el ensamblaje de polipéptidos que el gen mismo. Por esta razón, un ARNm también se puede describir como una cadena de "sentido" o un ARN de codificación. Los ARNm eucariotas no se sintetizan en su forma final (o madura) sino que, en su lugar, se deben tallar (o procesar) a partir de pre-ARNm mucho más grandes.

Figura 11.3. Estructura bidimensional de un ARN ribosómico bacteriano. Muestra el emparejamiento de bases extensivo entre las diferentes regiones de la cadena simple. La sección expandida muestra la secuencia de bases de un tallo y un bucle, que incluye un par de bases no estándar (G-U) y un nucleótido modificado, metiladenosina. Una de las hélices está sombreada de forma diferente porque juega un papel importante en la función de los ribosomas.
El uso de ARN mensajero permite a la célula separar el almacenamiento de información de la utilización de la información. Mientras que el gen permanece almacenado en el núcleo como parte de una enorme molécula de ADN estacionaria, su información se puede transmitir a un ácido nucleico móvil mucho más pequeño que pasa al citoplasma. Una vez en el citoplasma, el ARNm puede servir como plantilla para dirigir la incorporación de aminoácidos en un orden particular codificado por la secuencia de nucleótidos del ADN y ARNm. El uso de un ARN mensajero también permite que una célula amplifique en gran medida la emisión de polipéptidos. Una molécula de ADN puede servir como plantilla en la formación de muchas moléculas de ARNm, cada una de las cuales puede usarse en la formación de un gran número de cadenas de polipéptidos.
Las proteínas se sintetizan en el citoplasma mediante un complejo proceso llamado traducción. La traducción requiere la participación de docenas de diferentes componentes, incluidos los ribosomas. Los ribosomas son "máquinas" citoplásmicas complejas que se pueden programar, como una computadora, para traducir la información codificada por cualquier ARNm. Los ribosomas contienen proteínas y ARN. Los ARN de un ribosoma se llaman ARN ribosómicos (ARNr) y, al igual que los ARNm, cada uno se transcribe a partir de una de las cadenas de ADN de un gen. En lugar de funcionar como una entidad informática i digital, los rRNA proporcionan soporte estructural analógico y catalizan la reacción química en la que los aminoácidos se unen covalentemente entre sí. Los ARN de transferencia (o ARNt) constituyen una tercera clase importante de ARN que se requiere durante la síntesis de proteínas. Se requieren ARN de transferencia para traducir la información en el código de nucleótidos de ARNm en el "alfabeto" de aminoácidos de un polipéptido.
Tanto los rRNA como los tRNA deben su actividad a sus complejas estructuras secundarias y terciarias. A diferencia del ADN, que tiene una estructura helicoidal bicatenaria similar, independientemente de la fuente, muchos ARN se pliegan en una forma tridimensional compleja, que es marcadamente diferente de un tipo de ARN a otro. Por lo tanto, al igual que las proteínas, los ARN llevan a cabo una amplia gama de funciones debido a sus diferentes formas. Al igual que con las proteínas, el plegamiento de moléculas de ARN sigue ciertas reglas. Mientras que el plegamiento de la proteína es impulsado por la extracción de residuos hidrófobos en el interior, el plegamiento de ARN es impulsado por la formación de regiones que tienen pares de bases complementarias. Las regiones con pares de bases típicamente forman "vástagos" bicatenarios (y de doble hélice), que están conectados a "bucles" monocatenarios. A diferencia del ADN, que consiste exclusivamente en Watson-Crick estándar (GC, AT) pares de bases, los ARN a menudo contienen pares de bases no estándar y bases nitrogenadas modificadas. Estas regiones no ortodoxas de la molécula sirven como sitios de reconocimiento de proteínas y otros ARN, promueven el plegamiento de ARN y ayudan a estabilizar la estructura de la molécula. La importancia del apareamiento de bases complementario se extiende mucho más allá de la estructura de tRNA y rRNA. Como veremos a lo largo de este capítulo, el emparejamiento de bases entre las moléculas de ARN desempeña un papel central en la mayoría de las actividades en las que los ARN están comprometidos.
Los roles de mRNAs, rRNAs y tRNAs se exploran en detalle en las siguientes secciones de este capítulo. Las células eucariotas forman una serie de otros ARN, que también desempeñan papeles vitales en el metabolismo celular; estos incluyen ARN nucleares pequeños (snARN), ARN nucleolares pequeños (ARNs sno), ARN pequeños de interferencia (ARNip), ARNip, microARN (miARN) y una variedad de otros ARN no codificantes, es decir, ARN que no contienen información para aminoácidos. La mayoría de estos ARN cumple una función reguladora, controlando varios aspectos de la expresión génica, o incluso sirviendo como defensa contra infecciones virales. Varios de estos ARN han irrumpido en la escena en los últimos años, recordándonos una vez más que hay muchas actividades celulares ocultas esperando a ser descubiertas.
12. Síntesis de proteínas, transcripción
12. Síntesis de proteínas, transcripción
La transcripción es un proceso en el que una cadena de ADN proporciona la información para la síntesis de una cadena de ARN mensajero. Las enzimas responsables de la transcripción tanto en células procariotas como eucariotas se denominan ARN polimerasas dependientes de ADN, o simplemente ARN polimerasas. Estas enzimas son capaces de incorporar nucleótidos, uno a la vez, en una cadena de ARN cuya secuencia es complementaria a una de las cadenas de ADN, que sirve como plantilla.

Figura 12.1. Resumen de la transcripción del ARN (YouTube)
El primer paso en la síntesis de un ARN es la asociación de la polimerasa con la plantilla de ADN. Esto trae a colación una cuestión de interés más general, a saber, las interacciones específicas de dos macromoléculas, proteínas y ácidos nucleicos, químicamente muy diferentes. Del mismo modo que diferentes proteínas han evolucionado para unir diferentes tipos de sustratos y catalizar diferentes tipos de reacciones, también han evolucionado algunos de ellos para reconocer y unirse a secuencias específicas de nucleótidos en una cadena de ácido nucleico. El sitio en el ADN al que se une una molécula de ARN polimerasa antes de iniciar la transcripción se denomina promotor. Las ARN polimerasas celulares no son capaces de reconocer los promotores por sí mismas, pero requieren la ayuda de proteínas adicionales llamadas factores de transcripción. Además de proporcionar un sitio de unión para la polimerasa, el promotor contiene la información que determina cuál de las dos cadenas de ADN se transcribe y el sitio en el que comienza la transcripción. La ARN polimerasa se mueve a lo largo de la cadena de ADN molde hacia su extremo 5 (es decir, en una dirección de 3 a 5). A medida que la polimerasa progresa, el ADN se desenrolla temporalmente y la polimerasa ensambla una cadena complementaria de ARN que crece desde su extremo 5 en una dirección 3.
La ARN polimerasa cataliza la reacción altamente favorable en la que los sustratos de ribonucleósidos trifosfato (NTP) se escinden en nucleósidos monofosfatos a medida que se polimerizan en una cadena covalente. Las reacciones que conducen a la síntesis de ácidos nucleicos (y proteínas) son intrínsecamente diferentes de las del metabolismo intermediario. Considerando que algunas de las reacciones que conducen a la formación de moléculas pequeñas, como aminoácidos, pueden estar lo suficientemente cerca del equilibrio se puede medir una reacción inversa considerable, aquellas reacciones que conducen a la síntesis de ácidos nucleicos y proteínas deben ocurrir en condiciones en las que prácticamente no hay reacción inversa. Esta condición se cumple durante la transcripción con la ayuda de una segunda reacción favorable catalizada por una enzima diferente, una pirofosfatasa. En este caso, el pirofosfato (PPi) producido en la primera reacción se hidroliza a fosfato inorgánico (Pi). La hidrólisis del pirofosfato libera una gran cantidad de energía libre y hace que la reacción de incorporación de nucleótidos sea esencialmente irreversible.
A medida que la polimerasa se mueve a lo largo de la plantilla de ADN, incorpora nucleótidos complementarios en la cadena de ARN en crecimiento. Se incorpora un nucleótido en la cadena de ARN si es capaz de formar un par de bases adecuado (Watson-Crick) con el nucleótido en la cadena de ADN que se transcribe, solo cambia la timina por uracilo, pero sigue las mismas reglas de Chargaff. Una vez que la polimerasa ha pasado un determinado tramo de ADN, la doble hélice del ADN se re-forma a su condición de no transcripción. En consecuencia, la cadena de ARN no permanece asociada con su plantilla como un híbrido de ADN-ARN (excepto por aproximadamente nueve nucleótidos justo detrás del sitio donde opera la polimerasa). Las ARN polimerasas son capaces de incorporar de aproximadamente 20 a 50 nucleótidos en una molécula de ARN en crecimiento por segundo, y muchos genes en una célula se transcriben simultáneamente por cien o más polimerasas. La frecuencia con la que se transcribe un gen está estrechamente regulada y puede variar drásticamente dependiendo del gen dado y las condiciones prevalecientes.
Maduración del ARN mensajero

Figura 12.2. Editando el mensaje. Las proteínas funcionan como bloques ensamblables llamados dominios, los dominios pueden estar separados por secuencias que no van en la proteína final y deben ser eliminados por el empalme diferencial del ARN mensajero (YouTube). Se ha descubierto que un mensajero puede editarse alternativamente a varios mensajes maduros diferentes.
Mientras que la transcripción de genes codificadores de proteínas procariotas crea ARN mensajero (ARNm) que está listo para la traducción a proteínas, la transcripción de genes eucarióticos deja una transcripción primaria de ARN (pre-ARNm), que primero tiene que someterse a una serie de modificaciones para convertirse en un ARNm maduro.
Splicing o empalme diferencial
Una modificación muy importante del pre-ARNm eucariótico es el empalme de ARN. La mayoría de los pre-ARNm eucariotas consisten en segmentos alternantes llamados exones e intrones. Durante el proceso de empalme, un complejo catalítico de ARN-proteína conocido como espliceosoma cataliza dos reacciones de transesterificación, que eliminan un intrón y lo liberan en forma de estructura lariat, y luego unen los exones adyacentes.
Un intrón es cualquier secuencia de nucleótidos dentro de un gen que se elimina mediante corte y empalme de ARN durante la maduración del producto de ARN final. El término intrón se refiere tanto a la secuencia de ADN dentro de un gen como a la secuencia correspondiente en las transcripciones de ARN. Las secuencias que se unen en el ARN maduro final después del corte y empalme del ARN son exones. Los intrones se encuentran en los genes de la mayoría de los organismos y muchos virus, y pueden ubicarse en una amplia gama de genes, incluidos los que generan proteínas, en el ARN ribosómico (ARNr) y en el ARN de transferencia (ARNt). Cuando las proteínas se generan a partir de genes que contienen intrones, el empalme de ARN tiene lugar como parte de la ruta de procesamiento del ARN que sigue a la transcripción y precede a la traducción. La palabra intrón se deriva del término región intragénica, es decir, una región dentro de un gen.
Splicing o empalme alternativo
En ciertos casos, algunos intrones o exones pueden eliminarse o retenerse en ARNm maduro. Este llamado splicing alternativo y crea series de transcritos diferentes que se originan a partir de un único gen.

Figura 12.3. Sobrelapamiento de loci en un genoma. Diagrama de cajas/dominios de las proteínas del VIH, siendo estas: gag, pol, vif, vpr, vpu, tat, rev, env, y nef. Como se puede observar varios loci de genes se superponen en las mimas secuencias como en las proteínas gag y pol, en tat y rev o en env, tat y rev. Esta superposición permite almacenar más información genética en el mismo espacio.
Debido a que estas transcripciones se pueden traducir potencialmente a proteínas diferentes, el corte y empalme extiende la complejidad de la expresión del gen eucariótico. El procesamiento extenso del ARN puede ser una ventaja evolutiva hecha posible por el núcleo de eucariotas. En procariotas, la transcripción y la traducción suceden juntas, mientras que, en los eucariotas, la membrana nuclear separa los dos procesos, dando tiempo para que ocurra el procesamiento del ARN.
El empalme alternativo trae como consecuencia que un solo gen pueda almacenar información para más de una unidad funcional sea esta ARN o un polipéptido, incluso puede darse el caso de que dos loci almacenen entre los dos la información de un tercer loci fantasma como ocurre en el genoma de virus de ARN como el VIH causante del SIDA.
En eucariotas, el ARN mensajero maduro posterior a los empalmes debe exportarse al citoplasma desde el núcleo. Mientras que algunos ARN funcionan en el núcleo, muchos ARN se transportan a través de los poros nucleares hacia el citosol. En particular, esto incluye todos los tipos de ARN implicados en la síntesis de proteínas. En algunos casos, los ARN se transportan adicionalmente a una parte específica del citoplasma, como una sinapsis; luego son remolcados por proteínas motoras que se unen a través de proteínas enlazadoras a secuencias específicas (llamadas "códigos postales") en el ARN.
13. Síntesis de proteínas, traducción
13. Síntesis de proteínas, traducción
Para algunos ARN (ARN no codificante), el ARN maduro es el producto génico final. En el caso del ARN mensajero (ARNm), el ARN es un portador de información que codifica la síntesis de una o más proteínas de manera analógica. El ARNm que porta una única secuencia de proteína (común en eucariotas) es monocistrónico, mientras que el ARNm que porta múltiples secuencias de proteínas (común en procariotas) se conoce como policistrónico.

Figura 13.1. George Gamow y Marshall Nirenberg.
Cada ARNm consta de tres partes: una región 5 'no traducida (5'UTR), una región de codificación de proteínas o un marco de lectura abierto (ORF), y una región 3' no traducida (3'UTR). La región de codificación contiene información para la síntesis de proteínas codificada por el código genético para formar tripletes. Cada triplete de nucleótidos de la región codificante se denomina codón y corresponde a un sitio de unión complementario a un triplete anticodón en ARN de transferencia. Los ARN de transferencia con la misma secuencia de anticodón llevan siempre un tipo idéntico de aminoácido. Los aminoácidos se encadenan juntos por el ribosoma de acuerdo con el orden de tripletes en la región codificante. El ribosoma ayuda a transferir ARN para unirse al ARN mensajero y toma el aminoácido de cada ARN de transferencia y produce una proteína sin estructura. Cada molécula de ARNm se traduce en muchas moléculas de proteína, en promedio ~ 2800 en mamíferos.
Propiedades del código genético
Una vez que se reveló la estructura del ADN en 1953, se hizo evidente que la secuencia de aminoácidos en un polipéptido estaba especificada por la secuencia de nucleótidos en el ADN de un gen. Parecía muy poco probable que el ADN pudiera servir como una plantilla física directa para el ensamblaje de una proteína. En cambio, se supuso que la información almacenada en la secuencia de nucleótidos estaba presente en algún tipo de código genético. Con el descubrimiento del ARN mensajero como un intermediario en el flujo de información desde el ADN a la proteína, la atención se centró en la forma en que una secuencia escrita en un "alfabeto" de ribonucleótidos podría codificar una secuencia en un "alfabeto" que consta de aminoácidos.
Uno de los primeros modelos del código genético fue presentado por el físico George Gamow 1904-1968, quien propuso que cada aminoácido en un polipéptido estaba codificado por tres nucleótidos secuenciales. En otras palabras, las palabras de código, o codones, para aminoácidos eran tripletas de nucleótidos. Gamow llegó a esta conclusión con un poco de lógica de deductiva matemática. Él razonó que requeriría al menos tres nucleótidos para que cada aminoácido tuviera su propio codón único. Considere la cantidad de palabras que pueden escribirse usando un alfabeto que contiene cuatro letras diferentes que corresponden a las cuatro posibles bases que pueden estar presentes en un sitio particular en el ADN (o ARNm). Hay 4 palabras posibles de una letra 4=41; 16=42 palabras posibles de dos letras; y 64 =43 palabras posibles de tres letras. Debido a que hay 20 aminoácidos diferentes (palabras) que deben especificarse, los codones deben contener al menos 3 nucleótidos sucesivos (letras), evidentemente quedan varias sobrantes para sinónimos. La naturaleza de triplete del código fue pronto verificada en una serie de experimentos genéticos perspicaces conducidos por Francis Crick, Sydney Brenner y sus colegas de la Universidad de Cambridge.
Identificando los codones
En 1961, se conocían las propiedades generales del código, pero no se había descubierto una de las asignaciones de codificación de los tripletes específicos. En ese momento, la mayoría de los genetistas pensaban que tomaría muchos años descifrar todo el código. Pero Marshall Nirenberg 1927-2010 y Heinrich Matthaei 1929-vivo lograron un avance que utilizó una enzima polinucleótido fosforilasa para sintetizar sus propios mensajes genéticos artificiales y luego determinar qué tipo de proteína codificaron. Durante los siguientes cuatro años, varios laboratorios se unieron a la persecución y se construyeron ARNm sintéticos para probar las especificaciones de aminoácidos para los 64 posibles codones. El resultado fue el gráfico del decodificador universal para el código genético. El gráfico del decodificador enumera la secuencia de nucleótidos para cada uno de los 64 posibles codones en un ARNm.
El examen del diagrama de codones en la Figura 67 indica que las asignaciones de aminoácidos son claramente no aleatorias. Si uno busca las cajas de codones para un aminoácido específico, tienden a agruparse dentro de una porción particular de la tabla. La agrupación refleja la similitud en los codones que especifican el mismo aminoácido. Como resultado de esta similitud en la secuencia del codón, las mutaciones espontáneas que causan cambios de bases únicos en un gen a menudo no producirán un cambio en la secuencia de aminoácidos de la proteína correspondiente. Se dice que un cambio en la secuencia de nucleótidos que no afecta a la secuencia de aminoácidos es sinónimo, mientras que un cambio que causa una sustitución de aminoácidos se dice que es no sinónimo. Estos dos tipos de mutaciones describe varios de los diferentes tipos de mutaciones discutidas en este capítulo. Los cambios sinónimos tienen muchas menos probabilidades de modificar el fenotipo de un organismo que los cambios no anónimos. En consecuencia, es mucho más probable que los cambios no sinónimos sean seleccionados a favor o en contra por selección natural. Ahora que hemos secuenciado los genomas de organismos relacionados, como los de chimpancés y humanos, podemos observar directamente las secuencias de genes homólogos y ver cuántos cambios son sinónimos o no sinónimos. Es probable que los genes que poseen un exceso de sustituciones no sinónimas en sus regiones codificadoras hayan sido influidos por la selección natural.
El aspecto de "salvaguardia" del código va más allá de su degeneración. Las asignaciones de codones son tales que los aminoácidos similares tienden a ser especificados por codones similares. Por ejemplo, los codones de los diversos aminoácidos hidrofóbicos se agrupan en las dos primeras columnas del cuadro. En consecuencia, una mutación que da como resultado una sustitución de bases en uno de estos codones es probable que sustituya un residuo hidrófobo por otro. Además, las mayores similitudes entre los codones relacionados con aminoácidos se producen en los dos primeros nucleótidos del triplete, mientras que la mayor variabilidad se produce en el tercer nucleótido. Por ejemplo, la glicina está codificada por cuatro codones, todos los cuales comienzan con los nucleótidos GG. Una explicación para este fenómeno se revela en la siguiente sección en la que se describe el papel de los ARN de transferencia.
ARN de transferencia
Los ácidos nucleicos y las proteínas son como dos idiomas escritos con diferentes tipos de letras. Esta es la razón por la cual la síntesis de proteínas se denomina traducción. La traducción requiere que la información codificada en la secuencia de nucleótidos de un ARNm se decodifique y se use para dirigir el ensamblaje secuencial de aminoácidos en una cadena polipeptídica. La decodificación de la información en un ARNm se lleva a cabo mediante ARN de transferencia, que actúan como adaptadores. Por un lado, cada ARNt está unido a un aminoácido específico (como un ARNt aa), mientras que, por otro lado, ese mismo ARNt es capaz de reconocer un codón particular en el ARNm. La interacción entre codones sucesivos en el ARNm y aa-ARNt específicos conduce a la síntesis de un polipéptido con una secuencia ordenada de aminoácidos.

Figura 13.2. Código genético. Gráfico del decodificador universal para el código genético. El código es el mismo para todos los seres vivos, y le permite a los virus atacar a sus anfitriones.
Los ARN de transferencia traducen una secuencia de codones de ARNm en una secuencia de residuos de aminoácidos. La información contenida en el ARNm se decodifica a través de la formación de pares de bases entre secuencias complementarias en los ARN de transferencia y mensajero. Por lo tanto, al igual que con otros procesos que implican ácidos nucleicos, la complementariedad entre los pares de bases se encuentra en el corazón del proceso de traducción. La parte del ARNt que participa en esta interacción complementaria con el codón del ARNm es un tramo de tres nucleótidos secuenciales, llamado anticodón, que se encuentra en el ciclo medio de la molécula de ARNt. Este ciclo está compuesto invariablemente por siete nucleótidos, los tres medios constituyen el anticodón. El anticodón se encuentra en un extremo de la molécula de ARNt en forma de L opuesta al extremo al que está unido el aminoácido.
Acoplado de los aminoácidos en el polipetido
Es críticamente importante durante la síntesis de polipéptidos que cada molécula de ARN de transferencia se una al aminoácido correcto (afín). Los aminoácidos están unidos covalentemente a los 3 extremos de su (s) ARNt (s) relacionado (s) por una enzima llamada aminoacil-ARNt sintetasa (aaRS). Aunque hay muchas excepciones, los organismos típicamente contienen 20 aminoacil-tRNA sintetasas diferentes, una para cada uno de los 20 aminoácidos incorporados en las proteínas. Cada una de las sintetasas es capaz de "cargar" todos los ARNt que son apropiados para ese aminoácido.
Resumen del proceso de traducción
La traducción es el proceso que convierte de ARN mensajero a una proteína. El proceso es mediado por tres tipos de ARN. El ARN mensajero que sirve como mecanismo de transferencia de la información genética, el ARN ribosómicos que reconoce al ARN mensajero y lo encierra en una superestructura llamada ribosoma. Y el a de transferencia, que trasfiere un aminoácido por cada tres bases nitrogenadas. El tipo de aminoácido depende de la secuencia de las tres bases nitrogenadas llamadas codón. Este código ha sido resuelto y por lo general se encuentra expresada didácticamente en la tabla de codones. El ARN de transferencia posee un anticodón que reconoce a su codón, trasfiriendo un aminoácido a la cadena creciente de la proteína. La secuencia sigue creciendo hasta que se llega al codón de parada o hasta que se lee todo el ARN mensajero. Finalmente, la cadena de aminoácidos se enrosca sobre si misma por las interacciones moleculares formando una proteína.

Figura 13.3. Resumen de la síntesis de proteínas o traducción genética (YouTube) (YouTube)
Dogma central de la biología molecular
Aunque es un muy mal nombre, esta hipótesis ha sido reforzada por los descubrimientos en biología molecular, desde que fuera propuesta en los años 70s. En su forma más actual se puede definir cómo:
La información genética fluye desde los ácidos nucleicos hacia las proteínas y nunca en sentido inverso.
Esto implica que no existe un antiribosoma que pueda transformar la secuencia de aminoácidos en ARN mensajero. Esto explica la barrera de Weismann sobre la herencia lamarkiana. Si Lamarck hubiera estado en lo cierto, la célula debería contar con un antiribosoma que permita a una proteína modificada por el ambiente transferir ese cambio al ADN.
A pesar de lo tajante que puede parecer, el dogma central posee flexibilidades, por ejemplo, no dice nada sobre la direccionalidad del flujo de información entre los ácidos nucleicos. Aunque originalmente se consideraba que era direccional (ADN → ARN) la existencia de la enzima ARN transcriptasa reversa encontrada en retrovirus como el del VIH cambió todo. Posteriormente los retrotrasposones confirmaron lo que muchos sospechaban ya. El flujo de información genética entre el ADN y ARN es bidireccional (ADN ↔ ARN) a tal punto que el genoma humano ha sido esculpido por él. Existen más loci de retrotrasposones degenerados, que loci que contengan genes funcionales, por un margen muy amplio. La familia de loci originados por retrotrasposones más numerosa llamada L1 posee 500.000 copias, mientras que los loci codificantes son alrededor de 20.400. Aunque es verdad también que este mecanismo le permite a los virus infectar, también es uno de los mecanismos más importante para el desarrollo del potencial evolutivo de las especies.
14. Denaturación y renaturación del ADN
14. Denaturación y renaturación del ADN
El ADN es una molécula de tipo polímero, es decir una molécula compuesta por miles, millones o más átomos que se organizan
en módulos repetitivos en una estructura tridimensional que puede ser aproximada a modelos gracias a sus propiedades biofisicoquímicas. Pero el ADN es también una molécula encargada de almacenar información biológica, una propiedad mucho más difícil de describir, no solo en términos moleculares, sino también en términos filosóficos y ontológicos. Basados en información del grupo de Morgan, ya se sabía que el gen, fuera lo que fuere, se encontraba almacenado en segmentos de los cromosomas. Ya después de 1953 quedó establecido que la naturaleza química del gen se basaba en el ADN.

Figura 14.1. La mitosis. Texto La razón por la cual la meiosis funciona es porque al principio la célula posee cuatro copias del genoma, en la primera citocinesis los cromosomas duplicados se parten a la mitad, dejando las células en anafase II con dos copias del genoma. Al final de la última citocinesis los cromosomas homólogos se separan, dejando solo una copia del genoma por célula meiótica.
Por lo anterior se podía decir que la suma de todos los genes correspondería a la suma de las moléculas de ADN contenidas en los cromosomas. La totalidad de genes de un organismo se denomina genoma, y se encuentra contenido en una copia simple en una célula haploide de cromosomas simples. El genoma puede presentarse en copias en diferentes células. Una célula diploide que termina su mitosis posee dos copias del genoma; pero cuando va a indicar la mitosis, es decir, cuando tiene sus cromosomas duplicados, la célula diploide posee cuatro copias del genoma.

Figura 14.2. Efecto del calor en la estructura del ADN. Desde este punto en adelante modelamos al ADN del siguiente modo, una cinta que representa a los azucares y a los grupos fosfato, mientras que las bases nitrogenadas están representadas por escalones con sus colores estándar (YouTube).
A pesar de que inicialmente se comprendía que el genoma era algo complejo, con el paso de los años se ha llegado a comprender que la complejidad avizorada inicialmente era pobre con respecto a lo que se presenta en la naturaleza. Por ejemplo, inicialmente el genoma fue definido en torna a la suma de genes ubicados en las moléculas del ADN, esta visión se ha quedado corta al darse cuenta de que el ADN depende indisolublemente de proteínas que regulan su expresión o hasta su tasa de mutación. Sin embargo, antes de hablar de temas tan complejos, vale la pena iniciar con dos propiedades que afectarán al ADN, y son la denaturación y la renaturación.
La denaturación del ADN
Tal como fue propuesto originalmente por Watson, las dos hebras de la molécula del ADN se encuentran unidas entre sí por interaccione polares débiles, tanto por puentes de hidrogeno generadas por las bases nitrogenadas, así como por las interacciones de van de Waals que son importantes en moléculas de gran tamaño. La solución salina con ADN cambia sus propiedades físicas a medida que la molécula se denatura, por ejemplo, las medidas de absorbancia de luz ultravioleta. La temperatura para que la mitad de las hebras de ADN se hallan denaturado cambian dependiendo del contenido de bases nitrogenadas. Esto se debe a que la cantidad de puentes de hidrogeno es diferente, en la pareja A-T hay dos puentes de hidrogeno, mientras que en la CG hay tres. Si la molécula posee una mayor cantidad de parejas CG, la cantidad total de puentes de hidrógeno será mayor, y por lo tanto la molécula estará unida más fuerte y se necesitará una mayor temperatura para poder separar las dos hebras de la molécula del ADN.
La renaturación del ADN
La separación de las dos hebras de ADN mediante cambios de temperatura era un resultado esperable en base a lo que se sabe de las interacciones débiles, lo que en verdad llamó la atención de los bioquímicos fe que, al bajar la temperatura, las dos hebras se renaturen de forma determinista. La secuencia de bases nitrogenadas en cada hebra es complementaria a su contraparte, y las interacciones de todo el conjunto provocan que las dos hebras encajen perfectamente. Este resultado era prácticamente inconcebible en su tiempo.
En 1960, Julius Marmur "1926-1976" y Paul Doty "1920-2011" de la universidad de Harvard encontraron que al bajar la temperatura lentamente de una solución salina con ADN bacteriano que había sido denaturado térmicamente, este recuperaba todas las propiedades de doble hélice que había tenido antes de la denaturación. Esta propiedad de recuperar las propiedades originales ha servido de base para uno de los métodos de investigación más importantes de las ciencias biológicas modernas, y es la amplificación del ADN por medio de la reacción en cadena de la polimerasa que será descrita en artículos futuros con mayor detalle.
La renaturación es la propiedad del AND para recuperar sus propiedades originales después de haber sido denaturado, mientras que la recanalización hace referencia a la capacidad de la secuencia complementaria de detectar su contraparte y acoplarse a ella de forma específica sin la ayuda de enzimas. Esta propiedad ha permitido el desarrollo de múltiples métodos, por ejemplo, la detección de secuencias específicas mediante el uso de secuencias cortas de ADN de diferentes fuentes. Po ejemplo, sacar una secuencia de un gen de una mosca y buscar sus equivalentes en el ser humano. A esta metodología se la conoce como hibridación del ADN, y fue una de las técnicas más empleadas para el desarrollo de la taxonomía durante la segunda mitad del siglo XX.
Factores que afectan la denaturación y la renaturación
Si bien el genoma hace referencia a la totalidad del material genético, incluyendo los elementos regulatorios como las proteínas estructurales, en este contexto hace referencia solo al ADN y su longitud. Esto es porque la definición original del genoma estaba enmarcada únicamente en torno al ADN. Muchos factores determinan la tasa de renaturación de una preparación dada de ADN, por ejemplo:
(a) La fuerza iónica de la solución.
(b) Temperatura de incubación.
(c) La concentración de ADN disuelta en la solución
(d) Período de incubación
(e) El tamaño de las moléculas.
Tamaños de algunos genomas pequeños
Para determinar los efectos de diferentes variables se emplearon originalmente una serie de modelos biológicos con genomas relativamente pequeños, como el virus MS-2, el virus T4 y la bacteria Escherichia coli.
(a) MS-2: Es un bacteriófago “devorador de bacterias” icosaédrico de ARN de cadena positiva que infecta a la bacteria Escherichia coli: Es muy empleado en biología molecular.
(b) Virus T4. También es un bacteriófago de E. coli. Su estructura es compleja con cabeza, cuello y cola. Su ADN es bicatenario.
(c) Escherichia coli: Tal vez sea el ser vivo mejor conocido por la ciencia, siendo el modelo biológico más ampliamente empleado en el laboratorio.
En cuanto al tamaño respectivo de cada genoma:
(a) El virus MS-2 posee 4.0×103 pares de bases.
(b) El virus T4 posee 1.8×105 pares de bases.
(c) La bacteria E. coli posee 4.5×106 pares de bases.

Figura 14.3. Tamaño del genoma. En el eje (X) está tanto el tamaño como la velocidad de la reacción, y en el eje (Y) el porcentaje de asociación. Entre más pequeño es el genoma, la velocidad de asociación es mucho más rápida. En este caso el genoma del virus MS-02 se renatura antes que los demás.
Denaturación y renaturación del genoma viral y bacteriano
Comparando los genomas del virus MS, T4 y de la bacteria Escherichia coli la diferencia más evidente es su tamaño. Para comparar su renaturación es necesario que las moléculas que reacciones posean tamaños equivalentes, típicamente entre unos mil pares de bases. Las moléculas de ADN pueden ser fragmentados mediante una variedad de métodos, como el empleo de enzimas o forzar a la molécula a pasar por un filtro con altas presiones. Cuando se tienen estos tres tipos de genoma mezclados y cortados a 1000 pares de bases, el genoma del virus MS2 se renatura más rápido, seguido por el T4, y finalmente la de Escherichia coli.
Denaturación y renaturación del genoma eucariota
La cinética de renaturación de los genomas viral y bacteriano contrasta abruptamente con los resultados ara los genomas de eucariotas como los mamíferos, los cuales fueron obtenidos originalmente por Roy Britten y David Kohne de la Universidad de Carnegie. A diferencia de los genomas más simples, en los mamíferos se generan etapas marcadamente diferentes. Cuando la tasa de recanalización es mayor, significa que la probabilidad de que una secuencia encuentre su pareja es muchísimo mayor, mientras que cuando la velocidad es menor, la probabilidad de que una determinada secuencia encaje es menor.
Estas diferencias reflejan el hecho de que el genoma eucariota posee segmentos repetitivos y segmentos no repetitivos que operan casi al azar cuando tiene que ver con la recanalización de dos hebras de ADN. Este resultado fue la primera pista de que el ADN eucariota no era simplemente una progresión lineal de genes sin espacios intermedios, tal como si sucede en las bacterias. La complejidad del genoma eucariota es generalmente más grande.
15. Estructura del genoma
15. Estructura del genoma
Los estudios sobre la denaturación, la renaturación y sus velocidades permitieron identificar aspectos notables de la organización del genoma de los eucariotas. Cuando los fragmentos del ADN de plantas y animales son recanalizados, la curva típica muestra más o menos tres regiones que corresponde más o menos a tres diferentes tipos de secuencias de ADN. Las tres clases se recanalizan a velocidades diferentes, lo cual depende que tan al azar o que tan repetitiva es la secuencia, a mayor sea la repetitividad, más rápida es la recanalización. Las tres clases se denominan:
(a) Fracción altamente repetitiva
(b) Fracción moderadamente repetitiva
(c) Fracción no repetitiva
Fracción altamente repetitiva
En promedio constituyen entre 1% y el 10% de total de pres de bases del ADN. Las secuencias altamente repetitivas son por lo general muy cortas (las más grandes llegan a los cientos de pares de bases). Se encuentran agrupadas en nidos en los cuales una determinada secuencia se repite de forma cíclica persistente “tándem”. Las secuencias altamente repetitivas pueden clasificarse en varias categorías, incluyendo: ADN satélite, minisatélite, microsatélite. Estos nombres se originan a partir de propiedades físicas.

Figura 15.1. Los satélites al ADN principal. Cuando se centrifuga el ADN se forman varias bandas, una es la principal que es más densa, mientras que otras son más pequeñas y fueron denominadas satélites.
En promedio constituyen entre 1% y el 10% de total de pres de bases del ADN. Las secuencias altamente repetitivas son por lo general muy cortas (las más grandes llegan a los cientos de pares de bases). Se encuentran agrupadas en nidos en los cuales una determinada secuencia se repite de forma cíclica persistente “tándem”. Las secuencias altamente repetitivas pueden clasificarse e varias categorías, incluyendo: ADN satélite, minisatélite, microsatélite. Estos nombres se originan a partir de propiedades físicas.
El ADN satélite consiste en repeticiones entre cinco y algunos cientos de pares de bases, los cuales forman nidos “clusters” extremadamente largos, cada uno conteniendo millones de pares de bases. El nombre satélite emergió de una propiedad física que emerge de la alta repetición de estas secuencias en algunas especies.
ADN satélite
La composición entre el satélite y el ADN no repetitivo es tan diferente que al ser centrifugado los fragmentos se separan en bandas diferentes, el ADN satélite se ubica en una banda satélite con respecto al resto de gradiente de centrifugación. El ADN satélite tiende a acumular mutaciones rápidamente, lo que causa de las secuencias homólogas en una secuencia satélite cambien mucho aun en especies altamente relacionadas. Lo anterior permite realizar procesos taxonómicos en especies muy relacionadas.
ADN minisatélite
Las secuencias minisatélite rondan repeticiones entre 10 y 100 pares de bases agrupadas en nidos “clusters” de unas 3000 repeticiones. Por lo anterior, las secuencias minisatélite ocupan regiones del ADN considerablemente más pequeñas que las secuencias satélites, sin embargo, los minisatélites tienden a ser inestables, lo cual hace que varíen de una generación a la siguiente. En consecuencia, la longitud de un locus minisatélite individual es altamente variable en la población, incluso al interior de una misma familia. Debido a que son tan variables, una determinada cantidad de minisatélite pueden servir para la identificación inequívoca de un individuo con respecto a su propio material genético. Esto ha conllevado a que los minisatélites sean empleados como huellas dactilares genéticas “fingerprinting de ADN” en el contexto de las ciencias forenses.
ADN microsatélite
Los ADN microsatélite son las secuencias más cortas de todas las pertenecientes a la fracción altamente repetitiva. Típicamente son fragmentos de 1 a 9 pares de bases, y sus nidos “clusters” van de 10 a 40 repeticiones, los cuales se encuentran dispersos a través del genoma. Las enzimas especializadas en copiar al ADN tienen muchos problemas en copiar secuencias altamente repetitivas, lo cual conlleva a una tasa de mutaciones relativamente alta. Lo anterior implica que la longitud de un determinado microsatélite puede variar entre especies, variedades, e incluso hasta miembros de una misma familia. Dado lo anterior, los microsatélites se han convertido en la principal herramienta para el análisis de las poblaciones y las pruebas de identificación humana. El ADN microsatélite experimenta una variación lo suficientemente rápida como para poder realizar un análisis genealógico de poblaciones al interior de una misma especie, e incluso, algunos marcadores son tan mutables como para poder diferenciar hasta individuos al interior de una misma población. Otros estudios ya habían planteado la plausibilidad de que todos los humanos desandemos de una pequeña población africana que vivió hace unos 150 000-250 000 años.
En base a esto, es posible afirmar que las poblaciones africanas han tenido más tiempo para diversificarse genéticamente, mientras que solo una fracción decidió emigrar al norte y dar lugar a todas las demás poblaciones del mundo. Lo anterior implicaría que todas las poblaciones del planeta poseen solo una fracción de la diversidad de las poblaciones africanas. Otra conclusión algo paradójica es que, un africano puede llegar a parecerse genéticamente más a un europeo “en el caso que dicho africano pertenezca al pool genético del cual descienden los demás seres humanos del planeta” que con el vecino de una tribu cercana. En cualquier caso, estas conclusiones se basan en estudios cuantitativos realizados a través de marcadores como los minisatelites y microsatélites. En cualquier caso, desde la ciencia y las teorías evolutivas modernas, solo existe una raza de una sola especie humana viviendo en la actualidad, lo único que debe ser analizado cuantitativamente como un grupo cerrado relativamente es una población, pero las poblaciones pueden estar compuestas por una alta diversidad de grupos étnicos.
La mayor parte de los loci microsatélites se encuentran fuera de los genes, sin embargo, se han encontrado marcadores microsatélite dentro de genes e incluso asociados a patologías como el gen DM-1 responsable de la distrofia miotónica, en el cual los microsatélites con 50 o más repeticiones están asociados con la patología, mientras que los que presentan menos de 50 repeticiones presentan fenotipos normales. Una vez que se hizo evidente que los genomas eucariotas contienen grandes cantidades de repeticiones, los investigadores se interesaron por aprender donde era más probable encontrarlas al interior de los cromosomas. Gracias a los métodos de hibridación y al fenómeno de renaturalización del ADN ha sido posible concluir que la mayor parte de los microsatélites se ubican en los centrómeros de los cromosomas. Irónicamente, la función que originalmente se le había hipotetizado a todo el ADN por autores como Levene si es realizada por el ADN minisatélite y microsatélite, y es la de servir como un marco estructural para el genoma. Adicionalmente a esto, también puede llegar a cumplir roles de regulación o silenciamiento permanente de genes duplicados en ciertos casos.
Fracción moderadamente repetitiva
Las secuencias moderadamente repetitivas en las plantas y los animales pueden variar desde el 20% al 80% del genoma total de la especie. Esta fracción incluye secuencias que se repiten a través del genoma en cualquier parte desde unas pocas a unas decenas de miles de veces. Se incluyen en la fracción moderadamente repetitiva algunas secuencias que están involucradas con la expresión de ciertos genes, ya sea en términos de proteínas o ARN, así como aquellas que carecen de funciones codificantes, sea porque están involucradas en la regulación o porque en verdad no poseen función alguna.
Esta fracción incluye al ADN de genes que codifican para ARNs así como para ciertas proteínas regulatorias y estructurales importantes como las histonas. Las secuencias repetitivas que codifican para estos productos tándem a ser repetitivas y organizadas en tándem. Los genes que codifican para ARNs como los que componen al ribosoma deben estar repetidos múltiples veces debido a que estos no son amplificados posteriormente por el ribosoma. Aunque la producción de histonas si requiere de la amplificación de los ribosomas, se requiere muchísimas copias de estas proteínas durante el inicio del ciclo celular, que múltiples templados para su producción deben estar presentes de forma simultánea.
La mayor parte de las secuencias moderadamente repetitivas no poseen una función codificante, es decir, es incapaz de generar un producto génico sea ARN o proteínas debido a que no puede ser leído por las enzimas especializadas. A diferencia de las secuencias altamente repetitivas, las secuencias moderadamente repetitivas no se encuentran alojadas en nidos en tándem, por el contrario, se encuentran dispersas en toda la extensión del genoma. Esta fracción es dividida en dos grupos bastante famosos. El primer grupo se denomina SINE que significa elementos cortos intercalados “short interspersed elements” y el segundo grupo se denomina LINE que significa elementos largos intercalados “long interspersed elements”.
Fracción no repetitiva del ADN
Los genes mendelianos en la fracción de ADN no repetitiva
Como fue predicho por los mendelianos en los estudios sobre la genética clásica los genes se encuentran ubicados en una sola copia por cada segmento de un cromosoma simple. Usando una célula eucariota se encuentra en este estado se la denomina haploide, y por lo general corresponde a los gametos sexuales reproductivos. Los genes por lo general son secuencias no repetitivas, aunque existen excepciones, sin embargo, es posible afirmar que entre más lento es la recanalización de las dos hebras durante la renaturación menos repetitivo es el ADN. En ocasiones la recanalización es tan lenta en estas fracciones de ADN que puede presumirse que dichas secciones no son repetitivas en absoluto, en otras palabras, que los genes se encuentran en copias únicas en ciertas ocasiones. Estudios posteriores demostraron que los genes que provocaban la lentitud de la recanalización se comportaban de forma mendeliana, lo cual de por sí ya es una rareza.
La definición de gen y locus en una fracción de ADN no repetitiva
Estos genes que se encuentran en copias únicas y se comportan de forma mendeliana clásica nos permiten ingresar el concepto de gen y locus, por lo menos en sus definiciones más básicas. Un gen es una secuencia de ADN que almacena la información genética necesaria para la producción de otras moléculas como el ARN y las proteínas. El locus es la ubicación física del gen en un cromosoma, aunque parece redundante esta distinción obedece a un proceso histórico. Los genes fueron predichos matemáticamente antes de que su ubicación real al interior de los cromosomas fuera identificada gracias a los estudios del grupo de Morgan y colaboradores. Siendo una palabra latina, el plural de locus es loci. Sin embargo, los genes mendelianos son rarezas al interior del genoma, y nuevas propiedades estructurales del ADN han llevado a modificaciones de esta definición original.
Familias de genes y duplicaciones
Existen genes que, si se presentan en copias de sí mismos, y generan o herencias no mendelianas o rasgos diferentes o acoplados con sigo mismos. Estos genes copiados debido a su naturaleza misma pueden agruparse en un mismo grupo que posee una estructura muy similar. A estos grupos se los ha denominado familias de genes.
Virtualmente todos los genes existentes pueden ser agrupados en familias de genes, esto se debe a que los genes se producen más fácilmente de genes previos con leves modificaciones, que a partir de secuencias no codificantes. Estos últimos genes se denominan genes que han evolucionado de Novo, o lo que en términos pragmáticamente estadísticos podríamos decir, es como si evolucionaran de la nada o del caos.
Sin embargo, en cuanto a las familias de genes, existen algunas más famosas que otras, especialmente por presentarse copias al interior de un mismo genoma, este es el caso de las globinas, las actinas, las miosinas, los colágenos, las tubulinas, las integrinas, las proteasas de serina entre casi todas las demás proteínas eucariotas.
Tamaño de la fracción codificante con respecto a la no codificante del ADN
Ahora que el genoma humano ha sido secuenciado y después de más de una década de análisis es posible afirmar que la fracción de ADN codificante es increíblemente pequeña en comparación con el resto del genoma. De hecho, si esto se le hubiera sugerido a un genetista de la década de 1960, él hubiera considerado tal propuesta como ridícula. ¿Dada nuestra enorme complejidad cómo es posible que menos del 1,5% de nuestro genoma sea empleado para nuestra configuración corporal? Y aun así esa es la realidad que ha emergido de los estudios más modernos sobre la biología molecular y la genética.
ADN no codificante
Las secuencias de ADN no codificantes son componentes del ADN de un organismo que no codifican secuencias de proteínas. Aunque se podría pensar que se trata de ADN satélite y microsatélite la verdad es un poco más complicada, parte del ADN no codificante se transcribe en moléculas de ARN no codificantes funcionales (por ejemplo, ARN de transferencia, ARN ribosómico y ARN reguladores). Otras funciones del ADN no codificante incluyen la regulación transcripcional y traduccional de secuencias codificantes de proteínas, regiones de unión de andamios, orígenes de replicación de ADN, centrómeros y telómeros.
La cantidad de ADN no codificante varía mucho entre especies. A menudo, solo un pequeño porcentaje del genoma es responsable de codificar las proteínas, pero se muestra que un porcentaje creciente tiene funciones reguladoras. Cuando hay mucho ADN no codificante, una gran proporción parece no tener función biológica, como se predijo en la década de 1960. Desde entonces, esta porción no funcional “al menos para su individuo portador” ha sido controvertidamente llamada "ADN basura" (Pennisi, 2012).
El proyecto internacional Encyclopedia of DNA Elements (ENCODE) descubrió, mediante enfoques bioquímicos directos, que al menos el 80% del ADN genómico humano tiene actividad bioquímica (E. P. Consortium, 2012). Aunque esto no fue necesariamente inesperado debido a décadas anteriores de investigación que descubrieron muchas regiones no codificantes funcionales (Carey, 2015), algunos científicos criticaron la conclusión por combinar la actividad bioquímica con la función biológica, pues aunque se generen trascritos de ARN mensajero, muchos de estos no sirven para nada, siendo pseudogenes degenerados por mutaciones deletéreas (Maranda, Sunstrum, & Drouin, 2019; Xie, Chen, Xu, Zhao, & Zhang, 2019).
Las estimaciones para la fracción biológicamente funcional del genoma humano basadas en genómica comparativa oscilan entre 8 y 15% (Kellis et al., 2014). Sin embargo, otros han argumentado en contra de depender únicamente de estimaciones de genómica comparativa debido a su alcance limitado. Se ha descubierto que el ADN no codificante está involucrado en la actividad epigenética y redes complejas de interacciones genéticas y se está explorando en la biología evolutiva del desarrollo (Morris, 2012).
En la actualidad se distinguen las siguientes categorías de ADN no codificante.
ARN no codificante a proteína
Los ARN no codificantes son moléculas de ARN funcionales que no se traducen en proteínas. Los ejemplos de ARN no codificante incluyen ARN ribosómico, ARN de transferencia, ARN que interactúa con Piwi y los microARN. Se predice que los microARN controlan la actividad traduccional de aproximadamente el 30% de todos los genes que codifican proteínas en mamíferos y pueden ser componentes vitales en la progresión o el tratamiento de diversas enfermedades, incluido el cáncer, las enfermedades cardiovasculares y la respuesta del sistema inmunitario a la infección (Li et al., 2009).
Elementos cis y transregulatorios
Los elementos reguladores de cis son secuencias que controlan la transcripción de un gen cercano. Muchos de estos elementos están involucrados en la evolución y el control del desarrollo (Carroll, 2008). Los elementos cis pueden estar ubicados en regiones no traducidas de 5 'o 3' o dentro de intrones. Los elementos transreguladores controlan la transcripción de un gen distante. Los promotores facilitan la transcripción de un gen particular y están típicamente aguas arriba de la región de codificación. Las secuencias potenciadoras también pueden ejercer efectos muy distantes sobre los niveles de transcripción de los genes (Visel, Rubin, & Pennacchio, 2009).
Los intrones
Los intrones son secciones no codificantes de un gen, transcritas en la secuencia precursora de ARNm, pero finalmente eliminadas por empalme de ARN durante el procesamiento para madurar ARN mensajero. Muchos intrones parecen ser elementos genéticos móviles(Nielsen & Johansen, 2009). Los estudios de intrones del grupo I de los protozoos de Tetrahymena indican que algunos intrones parecen ser elementos genéticos egoístas, neutrales para el huésped porque se eliminan de los exones flanqueantes durante el procesamiento del ARN y no producen un sesgo de expresión entre alelos con y sin el intrón (Nielsen & Johansen, 2009). Algunos intrones parecen tener una función biológica significativa, posiblemente a través de la funcionalidad de la ribozima que puede regular la actividad de tRNA y rRNA, así como la expresión de genes que codifican proteínas, evidente en los huéspedes que se han vuelto dependientes de tales intrones durante largos períodos de tiempo; Por ejemplo, el intrón trnL se encuentra en todas las plantas verdes y parece haber sido heredado verticalmente durante varios miles de millones de años, incluidos más de mil millones de años dentro de los cloroplastos y otros 2-3 billones de años antes en los ancestros cianobacterianos de los cloroplastos (Nielsen & Johansen, 2009). Los intrones también son importantes para almacenar más de un gen en un mismo locus, por medio del proceso de maduración diferencial de un ARNm inmaduro (Hooper, 2014).
Los pseudogenes
Los pseudogenes son secuencias de ADN, relacionadas con genes conocidos, que han perdido su capacidad de codificación de proteínas o ya no se expresan en la célula. Los pseudogenes surgen de la retrotransposición o la duplicación genómica de genes funcionales, y se convierten en "fósiles genómicos" que no funcionan debido a mutaciones que impiden la transcripción del gen, como dentro de la región promotora del gen, o alteran fatalmente la traducción del gen, como codones de parada prematura o cambios de marco de lectura (Zheng et al., 2007). Los pseudogenes que resultan de la retrotransposición de un ARN intermedio se conocen como pseudogenes procesados; Los pseudogenes que surgen de los restos genómicos de genes duplicados o residuos de genes inactivados son pseudogenes no procesados (Zheng et al., 2007). Las transposiciones de genes mitocondriales que alguna vez fueron funcionales desde el citoplasma al núcleo, también conocidos como NUMT, también califican como un tipo de pseudogen común (Lopez, Yuhki, Masuda, Modi, & O’Brien, 1994). Los números ocurren en muchos taxones eucariotas. Si bien la Ley de Dollo sugiere que la pérdida de función en los pseudogenes es probablemente permanente, los genes silenciados en realidad pueden conservar la función durante varios millones de años y pueden "reactivarse" en secuencias codificantes de proteínas (Marshall, Raff, & Raff, 1994) y se transcribe activamente un número sustancial de pseudogenes (Tutar, 2012). Debido a que se presume que los pseudogenes cambian sin restricción evolutiva, pueden servir como un modelo útil del tipo y las frecuencias de varias mutaciones genéticas espontáneas (Petrov & Hartl, 2000).
Secuencias repetitivas de origen viral
Los transposones y retrotransposones son elementos genéticos móviles. Las secuencias repetidas de retrotransposón, que incluyen elementos nucleares intercalados largos (LINE) y elementos nucleares intercalados cortos (SINE), representan una gran proporción de las secuencias genómicas en muchas especies. Las secuencias Alu, clasificadas como un elemento nuclear corto intercalado, son los elementos móviles más abundantes en el genoma humano. Se han encontrado algunos ejemplos de SINE que ejercen control transcripcional de algunos genes que codifican proteínas.
Las secuencias endógenas de retrovirus son el producto de la transcripción inversa de genomas de retrovirus en genomas de células germinales. La mutación dentro de estas secuencias retro-transcritas puede inactivar el genoma viral. Más del 8% del genoma humano está formado por secuencias de retrovirus endógenas (en su mayoría descompuestas), como parte de la fracción de más del 42% que se deriva de los retrotransposones, mientras que otro 3% puede identificarse como restos de transposones de ADN. Se espera que gran parte de la mitad restante del genoma que actualmente no tiene un origen explicado haya encontrado su origen en elementos transponibles que estuvieron activos hace tanto tiempo (> 200 millones de años) que las mutaciones aleatorias los han vuelto irreconocibles (I. H. G. S. Consortium, 2001). La variación del tamaño del genoma en al menos dos tipos de plantas es principalmente el resultado de secuencias de retrotransposones (Hawkins, Kim, Nason, Wing, & Wendel, 2006).
Los telómeros
Los telómeros son regiones de ADN repetitivo al final de un cromosoma, que proporcionan protección contra el deterioro cromosómico durante la replicación del ADN. Estudios recientes han demostrado que los telómeros funcionan para ayudar en su propia estabilidad. El ARN que contiene repetición telomérica (TERRA) son transcripciones derivadas de los telómeros. Se ha demostrado que TERRA mantiene la actividad de la telomerasa y alarga los extremos de los cromosomas (Cusanelli & Chartrand, 2014).
16. Tipos de mutaciones
16. Tipos de mutaciones
De este punto en adelante los esquemas para la simbolización del genoma cambian, esto es debido a la extensión de los genes que, aunque en términos del genoma son pequeños, en términos absolutos de contar pares de bases pueden ser muy extensos. Las barras gruesas representan una sección de ADN de interés como un gen, pero también pueden representar loci no codificantes pero de interés como un marcador genético. Las barras delgadas representan también material genético pero que no es de interés para el contexto a analizar. En el genoma eucariota se encuentran secciones no codificantes y secciones codificantes y esta simbología permite representarlas rápidamente para poder exponer ciertas ideas como las que serán expuestas a continuación.

Figura 16.1. Modelo de barras. Modelo de barras, grueso para lo importante y delgado para lo no importante de una secuencia de ADN. La secuencia importante o resaltada puede ser un gen, un fragmento regulador o cualquier otra parte que sea el enfoque de la investigación.
Debido a que el ADN es el material genético tendemos a pensar en el como una molécula estática totalmente conservativa, que o no cambia o lo hace muy poco a través de la historia evolutiva –eso si es que considera la noción evolutiva en la biología, pero eso es mas de parte de los no-biólogos. El hecho ineludible es que el genoma es capaz de realizar cambios muy rápidos, no solo de una generación a la siguiente, sino al interior de un mismo individuo y más aun de una misma célula. Estos cambios pueden estar orquestados por mecanismos internos de origen interno “genéticos” o por la transducción de señales externas “epigenéticos”. En esta sección veremos como la biología ha adoptado una visión del mundo como la de Heráclito, todo está en movimiento, en un cambio constante, la estabilidad –en este caso de las especies y los genomas –es tan solo una apariencia. Evidentemente esta visión de trasfondo filosófico está reforzada por pruebas empíricas.
La primera teoría de la evolución tenía en su punto más débil una explicación lamarckiana de la genética, y aunque muchos digan que la selección natural es el motor de la evolución, hay que matizar mucho esa expresión. La selección natural es un mecanismo ecológico que elimina variantes, no las crea, por lo que en la base debía existir un mecanismo subyacente, el verdadero motor que pone en movimiento a la selección natural, aquel motor que arroja las variaciones que son susceptibles de ser seleccionadas. Años más tarde August Weismann refutaría la primera teoría de la evolución destruyendo su base lamarckiana, pero a pesar de que propuso una hipótesis genética de reemplazo llamada herencia dura, esta no pudo generar una explicación completa del problema. Posteriormente en 1916 Morgan y colaboradores dan soporte a las mutaciones como fenómeno que crea nuevos alelos, sin embargo, no sería hasta la elucidación de la maquinaria molecular y la naturaleza química de los genes que se pudo tener acceso al conocimiento específico de lo que son exactamente las mutaciones. En esta serie de artículos veremos la segunda parte de la Teoría Sintética de la Evolución, aquella que concierne no con la ecología y las poblaciones, sino aquella que concierne a la genética misma y al verdadero motor de la evolución, que es la mutación.
Mutaciones de un solo nucleótido o SNP
Literalmente mutaciones de nucleótido simple son las más comunes, pero también las menos importantes a la hora de explicar el origen de los genes, aunque sí que explican su diversificación secundaria. Una mutación de un solo nucleótido es un cambio en solo un par de bases nitrogenadas sea por inserción, deleción o cambio. Las inserciones son inocuas a menos que involucren la adición de un aminoácido crucial para la proteína, pero las deleciones e inserciones pueden ser particularmente nocivas pues alteran el marco de lectura de una proteína, lo cual la inhabilita funcionalmente. Los SNP constituyen hasta el 90% de todas las variaciones genómicas humanas, y aparecen cada 1,300 bases en promedio, a lo largo del genoma humano. Dos tercios de los SNP corresponden a la sustitución de una citosina (C) por una timina (T). Estas variaciones en la secuencia del ADN pueden afectar a la respuesta de los individuos a enfermedades, bacterias, virus, productos químicos, fármacos, entre muchos otros, lo cual en últimas conforma mucho del material base con el cual un individuo se enfrenta a la selección natural. Sin embargo, a pesar de ser tan comunes, los SNP nos dicen muy poco a cerca de la evolución a largo plazo de los seres vivos, y en especial en lo que respecta a una pregunta particularmente difícil si solo se conocen las mutaciones SNP, esa pregunta es ¿Cuál es el origen de los genes?
Mutaciones de genoma total o poliploidias

Figura 16.2. Las ranas poliploides. El poliploide es más grande, es una tendencia en las plantas (YouTube), pero que también se observa en las ranas del género Xenopus spp.
El tipo de mutación que explica las discrepancias con respecto a una cantidad doble de cromosomas en especies cercanamente emparentadas se denomina poliploidía. Una poliploidía se define como un cambio en el material genético en el cual el genoma completo de la especie es multiplicado por dos, si los parentales poseían dos cromosomas, los filiales poseerán cuatro cromosomas. La poliploidía emerge de un error en la meiosis, en la cual los cromosomas no se segregan a los diferentes gametos, y por lo tanto se producen la mitad de gametos sin cromosomas y la mitad de gametos con todos los cromosomas, por lo que se obtiene un gameto diploide. El punto es que se requiere de otro gameto diploide para que el poliploide se produzca y para esto existen dos opciones. La primera es que el gameto diploide sea ofrecido por una especie cercanamente emparentada también por un error en la meiosis. La segunda opción otro individuo de la misma especie, pero de genero diferente experimente el mismo error y ofrezca el gameto diploide complementario. Adicionalmente en los animales puede darse una poliploidía embrionaria en la cual los cromosomas que conforman al cigoto se duplican, pero no se segregan en células diferentes hasta que una segunda ronda de duplicación se da, por lo que en el primer clivaje se segregan el doble de cromosomas de la especie.
El fenómeno de la poliploidía es particularmente común en las plantas con flor, lo cual incluye a la mayoría de las especies domesticadas para la producción agrícola como el trigo, las bananas o el café. Algunas estimaciones afirman que más del 70% de las especies de plantas con flor son poliploides de otras. La duplicación de los cromosomas es un evento dramático para un individuo, el cual puede o no sobrevivir o experimentar cambios drásticos en el modo en que se relaciona con el ambiente. A simple vista muchos poliploides vegetales son más grades que sus parientes originales, pero al interior se empiezan a generar grandes cambios, como la tendencia a la eliminación de los genes redundantes, mediante el silenciamiento epigénetico o mutacional. El proceso de silenciado de genes es lento y afecta más a los genes de codifican a las proteínas que los genes regulatorios. Debido a su lentitud varios estudios han podido reportar la genética de este proceso en diversos estados de su historia evolutiva. De cualquier forma, las plantas con flor debido a su naturaleza poliploide tienden a poseer los porcentajes más altos de ADN codificante en comparación con los animales. Dado que las plantas con flor son en muchos casos hermafroditas, la producción de un solo individuo poliploide viable puede conllevar a la formación de una especie completa, lo cual es un ejemplo de especiación.
Duplicaciones parciales
La poliploidía es un caso extreme y dependiendo de la estructura genómica de la especie puede ser o muy común o muy rara. Según los datos recolectados es muy común para las plantas con flor, pero para los demás seres vivos es más bien un proceso extremo y raro.

Figura 16.3. Visualizando las duplicaciones. (A) En los chimpancés, por ejemplo, los loci para el gen de la amilasa AMY1 se encuentran en dos copias homólogas en cada uno de los cromosomas, como lo permite ver esta microfotografía empleando un marcador fluorescente para determinar la ubicación del gen. (B) En el humano el mismo gen se encuentra en copias variables al interior de cada cromosoma, aquí tenemos un cromosoma con 10 copias y otro con 4 copias del gen AMY1 de la amilasa.
Sin embargo, la poliploidía no es el único mecanismo que le permite a los seres vivos duplicar sus genes, existen formas en que pequeñas fracciones del genoma pueden duplicarse e insertarse de forma tal que adquieren los potenciales que otorgan la compensación de dosis genética. Estudios recienten han demostrado que las duplicaciones parciales de los genes suceden con una frecuencia sorprendentemente alta, lo cual no debería ser sorprendente, ya que la duplicación de los genes es la fuente principal de genes nuevos por medio de la subfuncionalización, la neofuncionalización y la fusión de dominios. Estos procesos son prácticamente uno de los principales motores del proceso evolutivo, ya que sin genes nuevos no habría posibilidad de cambio evolutivo.

Figura 16.4. Genes y proteínas. Los genes al expresarse forman las proteínas, las cuales son quienes realizan la mayor parte de las funciones en los seres vivos, la pérdida de un gen implica la pérdida de una proteína lo cual puede llegar a ser mortal para el individuo.
De acuerdo a un estimado reciente, cada gen en el genoma tiene alrededor de un 1% de probabilidad de ser duplicado cada millón de años, pero si usted posee unos 20 000 genes implica que cada millón de años 200 genes se habrán duplicado. La duplicación de los genes puede darse por múltiples mecanismos independientes a la poliploidía entre los que se puede contar los retrotrasposones, la transferencia viral y la recombinación no equivalente durante la profase I de la meiosis. Cabe añadir que no todos los genes tienen una misma tasa de duplicación, algunos tienden a duplicarse más que otros, lo cual implica que ciertos genes deben presentar una mayor cantidad de familiares en un mismo genoma.
De todos los mecanismos se cree que el más común es la recombinación no equivalente de los cromosomas durante la meiosis, lo cual generaría cromosomas más largos “con segmentos duplicados o inserción “y más cortos “con segmentos faltantes o deleción”. La recombinación no equivalente ocurre cuando los cromosomas que se encuentran en profase 1 no se alinean perfectamente. Los individuos que reciben los cromosomas con deleciones pueden sufrir patologías graves o incluso morir, pero en el caso de la inserción la compensación de dosis puede permitir al cromosoma nuevo no afectar mortalmente a su portador.
Otra consecuencia de esto, es que cuando el cromosoma alargado se recombine en la siguiente generación, los genes duplicados se intercambiarán en una cantidad diferente, o lo que es lo mismo, se genera una diversidad en los genes en la región afectada por la inserción, lo que conlleva a la generación de un nido de genes duplicados. Esta variación en el número de copias de un gen al interior de un nido “cluster” ha sido reportada para genes como el de la amilasa en el ser humano, en el cual se ha reportado variación entre 4 y 10 copias. Lo anterior implica que los cromosomas homólogos no necesariamente portan exactamente la misma información, tan solo requieren una integridad mínima para reconocerse mutuamente durante la meiosis, pero existe cierta flexibilidad en sus tamaños y contenidos. A esta tolerancia al cambio en sistemas complejos e integrados se la denomina resilencia.

Figura 16.5. Los genes son multi-modulares. Los genes pueden ser expresados en términos de módulos, sean estos repetitivos o no. Existe otro tipo de modularidad que se llama estructura cuaternaria, en el cual los módulos se encuentran separados como si fueran loci independientes.
Un pequeño porcentaje los genes que se encuentran en proceso de perderse en el caos del genoma no codificante adquieren mutaciones que otorgan ventajas a sus portadores mediante la adquisición de nuevas funciones. Cuando un gen lleva a cabo el proceso de neofuncionalización y subfuncioanlización apoyado por la compensación de dosis de una copia de respaldo se la denomina isoforma. Para ilustrar estos procesos analizaremos la familia de genes de las globinas, aunque ya lo hemos hecho con la cascada de coagulación
17. Efecto de la duplicación en el potencial evolutivo
17. Efecto de la duplicación en el potencial evolutivo
Aunque por lo general se nos dice que las proteínas tienen cuatro niveles de complejidad:
(a) Secuencia lineal o estructura primaria, esta se genera cuando el ribosoma la fábrica a partir de los mensajeros de ARN.
(b) La estructura secundaria es una conformación que adquiere la secuencia de aminoácidos cuando las interacciones moleculares empiezan a afectarlas generando hélices o láminas.
(c) La estructura terciara es una proteína funcional compuesta de hélices y láminas.

Figura 17.1. Defuncionalización deletérea. La defuncionalización es un proceso común, cuando un gen vital pierde su función, el indivioduo muere en el embrión o se genera una enfermedad en cualquier momento de la vida, disminuyendo la aptitud darwiniana del individuo.
Sin embargo, está visión es engañosa, entre la estructura secundaria y la terciaria podemos ubicar otro nivel de complejidad que podemos llamar dominios modulares. Una proteína funcional puede estar compuesta por varios dominios funcionales, los cuales le permiten operar en un determinado contexto.
Defuncionalización de un gen
La defuncionalización hace referencia a la pérdida total del gen, ya sea porque la proteína generada es no funcional –lo cual genera fenotipos recesivos. También puede darse el caso de que el mensajero de ARN al haber mutado no pueda ser leído por el ribosoma, o incluso que las enzimas que leen al ADN no puedan generar el mensajero de ARN, a esto se lo denomina cambio en el marco de lectura –efectos conocidos de mutaciones estilo SNP.
A parte de esto existen cambios epigenéticos que impiden que un gen duplicado se exprese, por ejemplo, las histonas que son proteínas estructurales del genoma pueden enroscarse en torno a las secciones duplicadas impidiendo su lectura por las enzimas especializadas. En otros casos, el gen mismo puede metilarse, lo cual impide que las enzimas puedan leerlo.

Figura 17.2. Defuncionalización no deletérea. Este es el destino más común para los genes duplicados, mientras que un locus es activo y evita que el individuo muera, con las generaciones el otro locus acumula mutaciones hasta perderse en el caos del ADN no codificante. Aun así, con las adecuadas técnicas de secuenciación es posible encontrar los rastros de estos genes perdidos o pseudogenes.

Figura 17.3. Subfuncionalización. La subfuincionalización, permite especializar los loci, lo cual puede ser conveniente a los seres vivos cuando se exponen a nuevos retos ambientales.
Compensación de dosis de un gen duplicado
Si dicho gen se perdiera por mutaciones del tipo SNP o por epi-alteraciones como el cambio de las histonas o la metilación del ADN entonces el organismo podría verse en graves riesgos de morir por defectos energéticos. Un gen duplicado permite que aun cuando uno de los nuevos se pierda, el locus ancestral –o el nuevo –seguirá funcionando, de forma tal que el organismo portador del locus alterado no sufre daños en sus sistemas metabólicos.
Subfuncionalización modular de un gen
La pérdida de la función de una proteína no necesariamente afecta a todos sus dominios funcionales, como mencionamos anteriormente, si al interior de un mismo gen se encuentran regiones funcionales, la pérdida de una de sus funciones puede conllevar a que dicho gen evolucione para realizar específicamente la función o funciones que le han quedado. Otro tipo de subfuncionalización puede abarcar la reducción de sustratos sobre los cuales actúa una proteína. En cualquier caso, todos estos fenómenos se basan en mutaciones de tipo SNP o en mutaciones en bucle o tándem.
Fusión de módulos
Bueno ¿y cómo aparece una proteína multimodular en primera instancia?, bueno esto no ocurre en las mutaciones de genoma completo, pero lo explicaremos aquí de todas formas. Por lo general los genes están flanqueados por secuencias regulatorias y un iniciador de la secuencia, así como un finalizador, el cual le dice a las enzimas especializadas –inicie a leer aquí y termine de leer aquí.

Figura 17.4. Fusión de módulos genéticos. Una vez que los dos módulos quedan conectados en un solo producto génico, uno de ellos puede acumular SNP sin riesgo de selección negativa para su portador. Con el tiempo uno de los módulos puede evolucionar a la perdida de la función, con lo que se convertiste en un elemento estructural de la proteína o a una nueva función, generando una proteína con múltiples dominios funcionales.
Si una mutación corte de forma tal que el segmento duplicado queda en tándem con respecto al viejo borrando la sección de terminación, la enzima leerá las dos secciones generando un único mensajero. Este mensajero único al ser leído por el ribosoma creará una proteína el doble de grande con dos dominios exactamente iguales. Lo cual permitirá que uno de ellos pueda evolucionar a funciones nuevas mientras que el viejo sigue su función normal.
Neofuncionalización de un gen duplicado
Un gen que está bajo compensación de dosis está libre de selección natural, lo cual le permite acumular rápidamente mutaciones por deriva genética. En muchos casos puede llevar a la perdida de la función y convertirse en un pseudogen, pero en otras puede ocurrir algo diferente.
La generación de funciones nuevas no necesita realmente grandes cambios, tal vez solo la alteración de una reacción química a otra muy relacionada altere la acumulación de un compuesto flexible por uno duro, en tal caso el organismo pasa de ser flexible y vulnerable, a inflexible pero muy duro. En cualquier caso, la nefuncionalización hace referencia a la adquisición de funciones aparentemente nuevas de un gen duplicado.

Figura 17.5. Neofuncionalización. Los pseudogenes al estar libres de la selección negativa pueden en ocasiones adquirir funciones nuevas que otorgan a sus portadores leves ventajas con respecto a sus competidores, lo cual los selecciona positivamente, permitiendo que acumulen mutaciones que filtradas por la selección conllevan a la formación de nuevas funciones y al nacimiento de nuevos genes.
La complejidad de los cambios genómicos
Evidentemente en el caso del cambio genómico una proteína puede tener simultáneamente en su historia duplicaciones, deleciones, subfuncionalizaciones solo para volver a duplicarse y quedar encadenada de nuevo formando nuevamente una proteína con más módulos.
Es por esta razón que el análisis evolutivo de las proteínas es tan complejo, pues pueden encontrarse rastros de módulos funcionales en proteínas que no se esperaría. Sin embargo, estos rastros conforman la base de una rica historia evolutiva que puede ser interpretada y que sirven como base para la generación de modelos evolutivos para sistemas que son muy complejos.
A pesar de lo complejo de este relato la propia secuenciación del ADN la ha corroborado, por todo el genoma se encuentran los restos de genes relacionados con otros funcionales, en el interior de genes funcionales se pueden encontrar restos de módulos no funcionales que sí lo son en otras proteínas.
18. Elementos móviles del genoma
18. Elementos móviles del genoma
Los elementos móviles del genoma son estructuras que pueden ser funcionales o no funcionales, pero que tienden a copiarse más de una vez en un determinado genoma. Debido a sus características de copia independiente del resto del genoma, se ha sugerido que estos provienen de virus que se han degenerado y que se han convertido en una parte fundamental del genoma para la generación de nueva información genética.
Evolución de los genes de la globina
La hemoglobina humana es un sistema relativamente complejo con cuatro unidades integradas, estas unidades son proteínas en sí mismas con una estructura terciara bien determinada. Por esta razón la hemoglobina humana se denomina una proteína con estructura cuaternaria. Cada unidad será denominada polipéptido y la hemoglobina humana posee cuatro. La estructura de cada globina cuando se analiza su gen productor siempre posee la misma estructura, tres exones y dos intrones. ¿Qué es un exón y que es un intrón? Bueno a pesar de que cubrimos parte de la estructura del genoma eucariota aun no los hemos mencionado.

Figura 18.1. Maduración del mensajero de ARN. Procesamiento del ARN mensajero, inicialmente cuando las enzimas crean el ARN lo leen como una sola entidad, pero posteriormente otras enzimas editan o maduran al ARN mensajero, transformándolo en el ARN mensajero maduro sin intrones.
Exones e intrones
Los genes no son exactamente como los simbolizamos. En la figura 1 se muestra el símbolo estándar para un gen, donde la barra gruesa a color simboliza el gen, mientras que la barra delgada simboliza el ADN no codificante. En realidad, la estructura se asemejaría más a esto (Figura 69) en la cual los genes se encuentran divididos por secuencias de ADN no codificante.

Figura 18.2. Estructura del gen de la globina.
Cuando las enzimas especializadas transforman el ADN a el ARN mensajero hay un paso de edición en la cual los intrones son editados del mensaje, quedando de esta forma un mensaje compuesto solo por algunos fragmentos originales. El fragmento de ADN que es eliminado por la edición intermedia en el mensajero de ADN se denomina intrón, mientras que la parte del mensaje que si es enviada al ribosoma para convertirse en proteínas se denomina exón.
Homólogos de las globinas
Como se mencionó anteriormente, aun cuando los genes al neofuncionalizar y subfuncionalizar alteran parte de su estructura, la mayor parte continua intacta, lo cual le permite a los biólogos buscar en los diferentes seres vivos miembros de una familia de genes determinada, y dependiendo de la cantidad de pares de bases que se asemejen pueden inferir el parentesco. Entre más antigua fue la separación de los genes más mutaciones han acumulado y por lo tanto más diferentes serán.
Cuando los científicos realizaron la búsqueda por las globinas se encontraron miembros de la familia realizando funciones en otros contextos de los animales, como por ejemplo las mioglobinas, pero también encontraron miembros de la familia en las plantas, ya que estas producen una proteína llamada le hemoglobina "portador de nitrógeno". En otros los casos no solo la secuencia en sí, sino la estructura misma del gen con tres exones dos intrones se mantenían constante, como el apellido de un clan.
Esto nos revela varias cosas, en primera instancia, las globinas como tal realizan funciones relacionadas con el transporte de electrones y la captura de oxígeno, dióxido de carbono y monóxido de carbono en diferentes contextos, y que como unidades independientes son completamente funcionales, lo cual va en contra del concepto de complejidad irreductible.
Buscando las primeras globinas
Si debemos creerle a Darwin, el origen de la hemoglobina debe encontrarse en un monómero, un solo fragmento que cumpla con la función. Evidentemente no podemos buscar en el sistema biológico humano debido a que en ese contexto se necesita la forma tetramérica, después de todo tanto el sistema como sus componentes evolucionan simultáneamente. Por lo anterior y debido a que las moléculas no se fosilizan se debe emplear la genética comparada. En este caso lo más objetivo es buscar en los familiares más lejanos, en el caso de los vertebrados, aquellos peces que se separaron del resto de los linajes antes de que evolucionaran las vértebras o las mandíbulas. Estos peces primitivos son los mixinos y las lampreas.
Al buscar en estos peces se encontró “oh sorpresa” que la hipótesis darwiniana queda soportada, en estos peces solo hay una copia del gen de la globina, lo que sugiere entre otras que los genes de la globina aparecieron por duplicación y que estos peses se separaron del resto antes de que esa duplicación diera lugar.
Duplicaciones y saltos, los genes no están quietos
Con la evolución de los peces vertebrados en algún punto el gen de la globina se duplicó, terminando la copia de respaldo cerca de la copia original en el mismo cromosoma. Sin embargo, los loci de las globinas son muy móviles y pueden trasladarse entre cromosomas. Por ejemplo, mientras que en los peces cebra y en las ranas del género Xenopus spp., ambos genes se encuentran en loci adyacentes. Por el contrario, en los vertebrados posteriores, uno de los genes de la globina saltó a otro cromosoma. Posteriormente, cada gen llevó a cabo una serie de duplicaciones y reorganizaciones generando los diversos tipos de globinas de los mamíferos actuales.

Figura 18.3. Esquema de la evolución de la familia de genes de la globina en los vertebrados. (1) simplificación de una globina ancestral, (2,3) duplicación del locus de la globina, (4,5) separación de los loci de las globinas en cromosomas independientes, (6,7) duplicaciones y reorganizaciones posteriores de los loci de las globinas.
Cuantas globinas tenemos los seres humanos
La hemoglobina del adulto está compuesta por cuatro polipéptidos de dos tipos, una alfa y la otra beta, pero esa no es toda la historia. Esto se debe a que los seres humanos poseemos otras copias del gen de la globina que permiten la producción de otros tipos de hemoglobina durante el desarrollo fetal. En el cromosoma 11 se encuentra la subfamilia de globinas beta, la cual se divide en globinas beta, gama y épsilon. Específicamente los loci son: globina beta, la globina delta, el pseudogen psi-beta, globina fetal gama A, globina fetal gama B, y globina embrionaria épsilon. En el cromosoma 16 se encuentra la subfamilia alfa, la cual se divide en las globinas alfa y zeta. Específicamente los loci son: globina alfa 1, globina alfa 2, pseudogen psi-alfa 1, pseudo gen psi-zeta y globina embrionaria zeta.
Esta familia de genes ilustra perfectamente el concepto de subfuncionalización mediante la especialización funcional. Cada una de las globinas de la familia alfa puede unirse con alguna de las beta, la hemoglobina resultante posee funciones específicas dependiendo del sistema en que se encuentre, sea este un embrión un feto o un adulto.
Pseudogenes en el genoma humano
En los nidos “clusters” de las globinas aparte de encontrar los genes de las cadenas extra que ya habían sido identificadas años atrás, los genetistas también se encontraron con loci fallidos, tal como se espera del proceso de duplicación, es decir la defuncionalización de un gen hasta convertirse en un pseudogen. Dicho de otra forma, un pseudogen es lo que queda de un gen antiguo que acumuló SNP de forma tal que en lugar de adquirir una función o especializarse perdió toda su funcionalidad. Tanto en el cromosoma 11 como en el 16 hay loci de pseudogenes de globinas.
El genoma humano posee 19 000 loci de pseudogenes, lo cual contrasta con los 20 500 loci para genes funcionales, es decir, casi la mitad de los se encuentran cargados de genes no funcionales! Sin embargo, que no codifiquen para proteínas no implica que no sirvan para nada, se han reportado pseudogenes que cumplen funciones de regulación.
Una de las características más sospechosas de las globinas es que en comparación con otros genes poseen una cantidad sorprendentemente pequeña de ADN no codificante, en otras palabras, el gen posee una integridad distintiva sin importar cuantas veces se hubiera copiado. Para repasar, las globinas parecen poder saltar de un lugar a otro manteniendo su estructura básica de forma intacta. O lo que es lo mismo, es como si el gen pudiera copiarse y viajar a otro cromosoma e insertarse de forma integral. El mecanismo que permite esta proeza genética es lo que permitió formular el concepto de genes saltarines o trasposones.
Genes saltarines
Si bien una parte integral de las explicaciones evolutivas recurren al azar, no todas las respuestas deben o pueden recurrir al azar debido a las probabilidades tan bajas. Por ejemplo, la probabilidad de que un loci evolucione para que su secuencia genética se parezca a la de otro gen es mucho más baja que la cantidad de electrones en el universo. Por esta razón se dice que la respuesta más parsimoniosa es que ambos genes posean un ancestro común, es decir que los dos genes con secuencias muy similares, pero con distintas funciones se originaron a partir de un gen ancestral. Hasta el momento hemos observado un mecanismo que permite la duplicación de genes y se denomina poliploidía. La poliploidía hace que se generen copias de un mismo gen en diferentes cromosomas, otro tipo de duplicación es la permitida por la recombinación durante la meiosis I. Sin embargo, existen duplicaciones que no encajan perfectamente con estos dos modelos mutacionales.

Figura 18.4. Barbara McClintock. Es la madre de la teoría de los genes saltarines (YouTube; YouTube), concepto que ha dado claridad a muchos procesos en genética evolutiva, y aplicaciones médicas como en el cáncer.
La hipótesis de los genes saltarines surgió como una forma de explicar cómo es que ocurrían genes duplicados en cromosomas diferentes, sin que estos tuvieran evidencia de una duplicación cromosómica mayor. Si se tratara de una poliploidía todo el cromosoma sería una copia homóloga de otro cromosoma al interior del mismo genoma, pero en estos casos lo único que es homólogo es una leve fracción, un locus con un cromosoma duplicado. La primera persona en sugerir que estos elementos se podían trasladar de un lugar al otro del genoma, es decir que podían saltar fue Barbara McClintock “1902-1992” trabajando en el laboratorio de Harbor en New York. El modelo biológico que permitió el desarrollo de este modelo fue el maíz.

Figura 18.5. Mecanismo de transposición. La transposición es el movimiento de un elemento móvil, el cual puede estar compuesto por varios loci así como elementos reguladores, de un lugar del genoma a otro
Los rasgos genéticos del maíz pueden revelarse en cambios de sus patrones en las hojas y en los patrones de coloración de las semillas. A finales de la década de los 40s Mc-Clintock encontró un grupo de mutaciones muy inestable que aparecían y desaparecían de una generación a la siguiente o inclusive durante la historia de vida de un solo individuo. Después de muchos años de estudio, ella concluyó que ciertos elementos genéticos pueden moverse de un cromosoma a otro. A estos elementos móviles ella los denominó Transponibles y a su organización transposón. Sin embargo, en la actualidad se usa el término transposón para designar al elemento móvil del ADN.
Simultáneamente, otros biólogos moleculares trabajando en bacterias no pudieron encontrar evidencias de estos elementos móviles. En sus estudios, los genes aparentaban ser elementos estables organizados en patrones lineares al interior del cromosoma circular, sin la presencia de secuencias no codificantes o pseudogenes. Los resultados de McClintock fueron ignorados hasta que a finales de la década de 1960 varios laboratorios encontraron se forma casi simultánea elementos móviles al interior del cromosoma circular de las bacterias. Fue en este marco de referencia que los elementos transponibles de McClintock pasaron a denominarse, transposones.
Estructura de un trasposón
Un transposón es un elemento que puede movilizarse al interior del genoma de los seres vivos, y al contrario de lo que puede pensarse involucra la presencia de varios loci. Generalmente lo que se percibe es el locus involucrado con rasgos en el fenotipo, pero al interior del transposón debe encontrarse otro locus que codifica para la enzima Transposasa.

Figura 18.6. Enzima de transposición. Una de las características de cualquier mutación es que es ciega, y la mayoría de las veces o causa enfermedades o es silenciada por el genoma. Cuando un transposón "morado" se inserta en medio de un gen "verde" puede causar enfermedades al destruir la función del gen. (YouTube).
Esta enzima puede literalmente catalizar el corte de todo el elemento móvil y pegarlo en otra posición del genoma. Esto implica que los genes lejos de ser elementos estructurales de un genoma, poseen una identidad propia, y pueden incluso llegar a comportarse como pequeños individuos que aportan a un todo, siendo capaces de moverse. Existe la posibilidad de que todo el elemento móvil pueda duplicarse parcialmente gracias a la recombinación no paralela en la meiosis I, lo cual genera nuevos transposones que a su vez pueden buscar otras posiciones del genoma.
Movimientos de un transposon en el genoma
La unidad a transponer aparte de codificar el locus de la transposasa y el locus que va a movilizarse, debe estar flanqueada por una secuencia repetitiva que marca la posición donde la transposasa debe pegarse. El primer paso es que la transposasa reconozca las secuencias de flanqueo y se pegue, luego la corta quedando una estructura con dos proteínas de transposasa en las puntas de una secuencia de ADN cortado. La transposasa posee otro dominio que le permite pegarse consigo misma, formando un elemento de ADN circular semejante a un plásmido que se movilizará al interior del genoma.
Una vez encontrado un nuevo lugar, el complejo de ADN-transposasa corta la hebra de ADN y media la inserción del elemento móvil. El proceso también involucra una duplicación pequeña del ADN blanco que queda flanqueando arriba y abajo al elemento móvil. Este tipo de duplicaciones sirve para identificar los loci de transposones al interior del genoma.
Efecto de los transposones en el genoma humano
Tal como lo demostró originalmente McClintock, el genoma de los eucariotas posee una cantidad relativamente alta de elementos móviles. De hecho, en el modelo biológico humano, cerca del 45% del ADN al interior del núcleo puede asociarse a elementos móviles. Esto implica que los elementos móviles tienden a perder su función, e incluso la capacidad de movilizarse una vez que han experimentado el cambio de lugar, lo cual va en concordancia con la naturaleza ciega de las mutaciones tipo SNP.
Al igual que ocurre con las duplicaciones del ADN total, cuando un transposón se moviliza de un lugar a otro tiende a ser silenciado por el genoma mediante cambios epigenéticos de las histonas o mediante metilaciones que bloquean su expresión. Peor aún, muchos elementos móviles tienden a insertarse en medio de loci de genes funcionales causando patologías metabólicas muy limitantes o inclusive fatales. Muchos casos de hemofilia se deben a este fenómeno. Se ha llegado a estimar que 1 de cada 500 mutaciones causantes de enfermedades se debe a un transposón que se insertó donde no debía. También se ha asociado que la activación de ciertos elementos móviles inactivados por las histonas o por metilaciones, pueden inducir al desarrollo de ciertos tipos de cáncer.
Los retrotrasposones
Los transposones al igual que cualquier gen deben venir de algún lugar, ¿cierto? Existe otro tipo de elemento móvil que permite la duplicación parcial de segmentos del ADN, a estos loci se los denomina retrotransposones. Un retrotransposón a diferencia de un transposón, se mueve al interior del genoma gracias a la maquinaria molecular normal, el loci completo se convertido a ARN mensajero “por lo que la copia original queda intacta”, pero en esta posición, existe una proteína llamada ARN transcriptasa reversa que transforma al ARN mensajero a ADN creando una copia del gen, la cual procede a insertarse en otro lugar. Muchos retrovirus poseen la misma enzima como los del VIH.
La importancia de los virus en la evolución
Familias de genes como los de las globinas pueden ser explicadas como transposones y retrotransposones. La retrotrasnposición da origen a la copia del gen, sin que se requiera que todo el genoma se duplique. Posteriormente uno de los locus puede transponerse a otro cromosoma, y allí volver a copiarse por retrotransposición. Esto convierte a los genes que pueden copiarse y moverse en entidades con cierta individualidad, de hecho, en las globinas dicha individualidad puede verse en que todos esos loci mantienen una estructura común.
El hecho de que muchos virus denominados retrovirus puedan realizar proezas semejantes ha permitido generar hipótesis sobre la importancia de los virus en la evolución biológica, ya que sin la enzima transcriptasa reversa, muchas familias de genes no hubieran podido surgir. Lo anterior implica que al menos los retrovirus al donar la enzima transcriptasa reversa han servido como parte del
verdadero motor evolutivo que es la variación aleatoria,
ya que, al permitir una copia de genes más fácil, aceleran el potencial para la creación de nuevos rasgos al interior de los seres vivos. Personalmente, a diferencia de lo que proponen autores como Máximo Sandin, esto es perfectamente concordante con el esquema de la síntesis evolutiva moderna, después de todo los loci nuevos creados por la retrotransposición deben medirse ante la selección natural cuando subfuncionalizan o neofuncionalizan.
Cuantificación de los retrotrasposones
Las secuencias moderadamente repetitivas pueden componer aproximadamente el 80% del genoma de un eucariota y la mayoría de ellas pertenecen a familias de pseudogenes relacionables a retrotransposones. De hecho, las dos familias de secuencias moderadamente repetitivas más importantes denominadas Alu y L1 son retrotransposones. Los L1 por ejemplo, poseen un locus que codifica para una proteína con dos dominios, una transcriptasa reversa que transforma mensajeros de ARN a ADN, y al mismo tiempo posee un dominio endonucleasa que permite a ese ADN insertarse en el genoma. Lo interesante es que los humanos poseemos 500 000 copias de ese gen, aunque la mayoría de ellas se encuentra dañada debido a mutaciones de tipo SNP. Ese valor de 500 000 copias contrasta con los 20.400 loci de genes que expresan proteínas.
ADN basura
Cuando algo de este tamaño aparece en el genoma de un organismo, la primera pregunta que viene a la mente s ¿cuál es su función? Muchos biólogos moleculares pensaban que las secuencias moderadamente repetitivas eran principalmente basura, de acuerdo con este punto de vista, los retrotransposones son una especie simbionte comensalista a nivel molecular, que, a diferencia de un virus, no mata, pero tampoco beneficia a su portador. Aunque este pudiera ser su origen, o incluso asumiendo que los primeros retrotransposones fueran virus degenerados, con un ciclo de infección reducido, no implica que no posean función. El punto es que, aunque no posea una función para el individuo, los retrotransposones si constituyen una fuente de variabilidad y potencial evolutivo para linajes de seres vivos.
Los elementos móviles pueden en ocasiones arrastrar partes del genoma del hospedero moviéndolos de un lugar a otro. Este mecanismo cuando es muy limitado puede permitir el traslado de dominios o módulos de un gen a otro gen, lo que genera proteínas multimodulares nuevas. Cuando es más extenso puede duplicar uno o más genes. Este proceso es facilitado en el genoma eucariota debido a los intrones, si el segmento nuevo se inserta en un intrón, no se dañará la proteína generada por el gen al cual se le ha adicionado el nuevo elemento. Muchas proteínas, como las que están relacionadas con el flagelo bacteriano manifiestan evidencia de haber surgido por este mecanismo.
Las secuencias que emplean algunos elementos móviles para moverse de un lugar a otro, han sido asociadas a los elementos reguladores del ADN. En otras palabras, las secuencias que regulan la expresión de las proteínas en el genoma eucariota tienen su origen en las secuencias que permiten la transposición de los transposones. Adicional a lo anterior, muchos elementos móviles que carecen de función poseen similitudes de secuencia con genes funcionales en otros vertebrados, lo cual convierte al ADN basura en un registro fósil de nivel molecular.
En algunos casos, los elementos móviles poseen relaciones con genes activos y con pseudogenes. La enzima telomerasa, la cual juega un papel preponderante en la replicación final del ADN pudo haberse originado por retrotranposición de un elemento móvil ancestral. La enzima que media la reorganización de las regiones hipervariables de los anticuerpos en el sistema inmune también posee rastros de ser un retrotransposon. La misma familia de las globinas muestra evidencias simultáneas de transposición y retrotransposición.
Un punto es claro, la retrontransposición y la transposición poseen un efecto profundo en el genoma de los eucariotas y procariotas. Resulta interesante ver que unas décadas atrás la mayoría de los biólogos moleculares concebía al genoma como una entidad muy fija y de difícil cambio, lo cual complicaba algunos aspectos de la teoría de la evolución.
De hecho, la propuesta original de la TSE se basaba en que el genoma solo podía experimentar cambios muy leves de una generación a otra, pues de lo contrario, su estabilidad general se vería muy afectada. Esto a su vez concluyó en la propuesta del gradualismo filético a ultranza, o lo que es lo mismo, concebir a la evolución como un proceso absolutamente gradual. Estos descubrimientos han demostrado que el genoma posee una flexibilidad y resiliencia muy grandes, lo cual facilita explicaciones evolutivas, en otras palabras, el potencial evolutivo de los seres vivos es mayor de lo que se habría esperado originalmente. Gracias a estos descubrimientos en 1983 se le otorgó a Barbara McClintock el Premio Nobel.
Genes de novo
La respuesta a la pregunta ¿de dónde surgen los genes? Parecía muy clara cuando comenzó en nuevo milenio, y la respuesta era, por duplicación de genes previos. Esto es porque aún el biólogo que confiaba más en el azar vería muy improbable que los genes pudiesen evolucionar por SNP de secuencias no codificantes sin regiones promotoras o reguladoras.
Sin embargo en 2006 algo sucedió que estremeció los fundamentos de la biología molecular realizado por Yuuki Hayashi, Takuyo Aita, Hitoshi Toyota, Yuzuru Husimi, Itaru Urabe y Tetsuya Yomo (Hayashi et al., 2006) en el cual demuestran experimentalmente el potencial de un gen de emerger del caos del ADN no codificante. Debido a la imposibilidad teórica, el hecho de que suceda en la práctica demuestra como los procesos de variación aleatoria SNP y la selección natural son capaces de crear información prácticamente de la nada.
A este tipo de genes que emergen de mutaciones aleatorias sin un gen previo que sirva de base se los denomina genes de Novo y desde entonces han sido reportados incluso en el genoma humano.
Importancia evolutiva de los genes duplicados
Los genes nuevos son las principales fuentes de innovación en los genomas. Sin embargo, hasta años recientes el entendimiento de como los genes nuevos se originaban y como evolucionaban una vez habían emergido brillaba por la ausencia de apropiadas bases de datos moleculares “y muchos lo siguen pensando así”. Con el advenimiento de la era genómica llegó una revolución en la información disponible para estudiar genes nuevos. Por primera vez, décadas de principios teóricos pueden ser medidos de manera empírica, junto con nuevas líneas de investigación Aunque la totalidad del artículo en que se basa la anterior introducción será sujeto de un análisis futuro, en este punto me enfocaré únicamente en tres tópicos de manera muy muy resumida, la duplicación de genes, la subfuncionalización y más importante aún, la neofuncionalización (Cardoso-Moreira & Long, 2012). Aunque existen otras formas para la aparición de genes nuevos, el asunto empírico es que muchos de los genes de los sistemas complejos de múltiples componentes integrados que interactúan entre si poseen líneas de evidencia que los implica como genes duplicados que han experimentado subfuncionalización y neofuncionalizacion. Es importante recalcar que, si bien la primera parte de la explicación para los sistemas complejos es la duplicación, la actual estrategia del DI como bien lo expone Behe (William & Ruse, 2004) es básicamente negar la otra mitad de la historia, la neofuncionalización y la subfuncionalización.
Durante la duplicación de genes, tenemos una secuencia de ADN que al pasar por una mutación se duplica en una misma célula y luego esa duplicación es mantenida en el resto de los descendientes. En tal caso, hay que distinguir entre dos tipos diferentes, la duplicación a gran escala de genomas completos denominada poliploidia de la duplicación de genes o secuencias de ADN más pequeñas (Conant & Wolfe, 2008). Un detalle a rescatar es que, la historia subsecuente de un gen duplicado “la cual parecen querer olvidar, los defensores del DI y del creacionismo” es que dependerá de qué tipo de evento duplicador la hubiera generado, es decir, los genes que se fijen y su historia diferirá de si nacieron como consecuencia de una duplicación a pequeña escala o por un evento de poliploidia (Conant & Wolfe, 2008; Maere et al., 2005; Wapinski, Pfeffer, Friedman, & Regev, 2007). Los genes duplicados representan una porción bastante grande de los genomas de diferentes tipos de eucariontes, por ejemplo, el 17% en algunas bacterias, al 65% en la planta Arabidopsis (Zhang, 2003). La diversidad de las duplicaciones es amplia y no se restringe a los polos opuestos de, un solo gen o el genoma completo, y de hecho es bastante común que se den duplicaciones de grupos de genes (Cardoso-Moreira & Long, 2012).
Un detalle interesante sobre la determinación de la duplicación de genes, resulta de cumplir a cabalidad las hipótesis originales de Darwin. A medida que los duplicados son más viejos y han tenido más tiempo para mutar, estos se hacen más diferentes, mientras que los duplicados más jóvenes son más semejantes entre sí. El problema de tener genes duplicados muy semejantes, es que los métodos moleculares actuales los hacen colapsar cuando se ensamblan los genomas completos, y por lo tanto los hacen pasar como si fueran un solo gen. Como resultado, el número de genes duplicados recientemente es mucho más alto de lo que puede llegar a medirse (Bailey et al., 2002). No nos enfocaremos en los mecanismos moleculares de las duplicaciones ya que los proponentes de DI parecen no cuestionar la existencia de la duplicación.
Ahora, digamos que, en lugar de tener un gen con una función específica, afinada, especifica y completamente irrenunciable llamada A, después del evento duplicador tenemos dos genes que expresan la misma condenada proteína. ¿Que pasara? Aparentemente según los escritores del diseño inteligente, no mucho, ya que, para ellos, un gen duplicado es solo información redundante. Una vez se ha duplicado un gen ocurre el fenómeno clásico denominado compensación de dosis. Y digo clásico porque su efecto es demostrable aun desde los primeros trabajos en genética, con la genética clásica mendeliana. La compensación de dosis se presenta cuando se tienen dos que generan la misma proteína, cuando ello ocurre, solo se requieren de uno de los para generar la cantidad de proteína necesaria para llevar a cabo la función. En tales casos, se puede tener un gen recesivo que no cumple la labor, ya que en tal caso el dominante compensará la dosis produciendo la misma cantidad de proteína y generando el fenotipo normal.
Los genes duplicadores experimentan la misma cuestión, en el momento en que los genes se duplican, uno compensará la necesidad del otro, por lo que, aun cuando la función A sea completamente irrenunciable, esta podrá llevarse a cabo por la presencia de uno de los dos loci, mientras que el otro podrá acumular mutaciones. La acumulación de mutaciones conllevará a tres destinos posibles, “la neutralización perdida de función o pseudogenización; la subfuncionalización y la neofuncionalización” (Cardoso-Moreira & Long, 2012). Dada la condenada aleatoriedad de las mutaciones, la ruta más común para el gen duplicado es la pseudogenización, lo que implica la perdida de la función del gen, lo que lo hace ingresar en la categoría de genes no codificantes, o bajo el infortunado nombre de ADN basura. El asunto puede ser tan severo, que se ha estimado que existe al menos un pseudo gen por cada ocho genes en el gusano C. elegans (Harrison, Echols, & Gerstein, 2001), y para los humanos, “cuya cascada de coagulación intentamos explicar” la cantidad de pseudogenes es de uno por cada dos genes funcionales (Harrison et al., 2001).
Es importante resaltar que, no todos los pseudogenes se generan por duplicaciones, muchos pseudogenes han aparecido por la pérdida de presiones de selección, por ejemplo, las pérdidas de la sensibilidad del olfato en muchos primates han dado origen a la pseudogenizacion de los genes responsables por un buen olfato (Rouquier, Blancher, & Giorgi, 2000). El asunto es que teóricamente, el resultado más probable de la vasta mayoría de mutaciones es el de mutaciones deletéreas, o neutrales. Pero aun así… Las duplicaciones pueden generar neofuncionalizaciones, e modelo fue ya propuesto por Ohno bajo el argumento de la compensación de dosis (Ohno, 1970). Lo interesante de la selección natural es que actúa como un filtro amplificador o un trinquete irreversible; una vez que ocurre una rara mutación ventajosa, esta es fijada y amplificada rápidamente en el interior de la población (Cardoso-Moreira & Long, 2012). Un ejemplo, ahora clásico de nefuncionalización ocurrió en una especie de monos vegetarianos, en la cual se duplicó el gen de la ribonucleasa. Después de la duplicación, el gen duplicado sufrió una serie de mutaciones que conllevaron a generar una función digestiva más efectiva en un nuevo microambiente (Zhang, Zhang, & Rosenberg, 2002)
Recientes datos genéticos sugieren que las nuevas funciones pueden darse de manera más común, y aún más, que pueden emerger al mismo tiempo en que nace el gen duplicado. De hecho, resulta interesante que el único tipo de mutaciones duplicantes que dejan intactas a los genes es la poliploidia, los demás mecanismos de duplicación alteran de alguna forma a los genes durante el proceso “duplicaciones parciales, fisiones de genes, cambio de marco de lectura de exones, retrotrasposones, y la más rara de todas las rarezas teóricas genes DE NOVO” (Cardoso-Moreira & Long, 2012). Finalmente hay que tener en cuenta a la epigenética; aun cuando la secuencia genética del locus nuevo es igual, el mero hecho de encontrarse en otro lugar con diferente microambiente de cromatina es suficiente para desatar una nueva función (Cardoso-Moreira & Long, 2012).
Bueno ya esto se extendió mucho y tratar el tema completo requeriría su propia serie de artículos, así que resumiré brevemente la subfubncionalización. En ocasiones la proteína original lleva a cabo dos funciones de manera rudimentaria, la subfuncionalización le permite a los loci duplicados especializarse en una función diferente, los detalles los pueden leer en (Cardoso-Moreira & Long, 2012). En resumen, la duplicación no solo genera información redundante, es más, cuando se trata de duplicaciones internas sin alterar el tamaño del genoma de manera perceptible, las duplicaciones frecuentemente alteran al nuevo gen a formarse, lo cual genera directamente una nueva proteína. Que la nueva función de la proteína tenga una función o no es ya cuestión de suerte, claro, a menos “y en mi opinión” que se encuentren más mecanismos que limiten/potencien aún más la aleatoriedad en las mutaciones según sea necesario.
19. Identificación humana
19. Identificación humana
Podríamos escribir mucho sobre la matemática de la genética-evolución modernas, sin embargo, para los propósitos de este curso introductorio a la genética moderna nos enfocaremos en la identificación humana. La identificación humana ha sido uno de los problemas más grandes que han tenido que ir solucionando las ciencias forenses con el paso de los años. En términos prácticos la identificación humana forense se realiza para el caso en que no pueden emplearse otros medios como la identificación de rostros por medio de testigos. Casos donde la identificación humana es importante son: pruebas de parentesco como las de paternidad y maternidad; e identificación de material biológico como en violaciones o asesinatos. En las pruebas de paternidad, por ejemplo, se puede tener uno o varios padres posibles, sobre los cuales hay que realizar una decisión de tipo pecuniario.
En las pruebas de paternidad se puede tener el caso de bebes que han sido mal identificados al nacer, o de hijos perdidos por historias familiares, entre los que cabe destacar el caso de Emmanuel en Colombia (Enlace). La identificación humana directa por el contrario trata de establecer la fuente de material biológico como sangre o semen en casos de violaciones o para establecer la presencia de algún sospechoso en una escena del crimen. La identificación humana por medio de la genética se ha convertido en una de las herramientas más poderosas de los detectives a la hora de resolver casos complicados. En esta serie de artículos discutiremos algunos principios que rigen dicha metodología.
El principio de la identificación humana
Cuando llenamos un pedigrí en ocasiones realizamos un razonamiento que nos permite excluir o incluir un genotipo dado. Para emplear los razonamientos de la genética, primero debemos tener en cuenta el tipo de sistema que vamos a enfrentar. Por lo general podemos tener dos clases generales con algunas variaciones.
(a) Identificación de material biológico con respecto a una fuente, en este caso debemos analizar si un material le pertenece a un individuo dado al interior de una población completa, esto implica que el genotipo del material genético debe coincidir en un 100% con el individuo analizado.
(b) Pruebas de paternidad y otros grados de filialidad o parentesco, en este caso la similitud genética es inferior, por ejemplo, cada padre compartirá solo el 50% de los marcadores, a menos que los padres compartan los mismos alelos para un locus determinado.
El índice de similitud desciende, para dos hermanos dicigóticos y hermanos normales es del 50%, para gemelos idénticos es del 100%, los tíos con sus sobrinos comparten un 25% al igual que los abuelos, mientras que un primo en primer grado solo comparte el 12.5%. Esta similitud decreciente puede acarrear problemas a la hora de poder identificar o por lo menos excluir posibles parientes cercanos.
Inducción de genoma parental.
La inducción del genoma parental es un razonamiento que se basa en los genomas de los descendientes, para el cual generalmente se emplea análisis de pedigrí. En este caso podemos decir que el segundo alelo en cada uno de los parentales es recesivo.
Siendo los genomas de dos parentales: Ax × Ax
Y el genoma de una cría: aa
Podemos inferior que el alelo desconocido es un allo recesivo sin la necesidad de hacer pruebas de laboratorio.
Fuentes de marcadores para la identificación humana
Existen formas de evadir el problema de la similitud por segregación, por ejemplo, existen fuentes que no segregan o recombinan entre géneros, lo que da acceso a similitudes del 100%. Para el caso de las madres es el ADN mitocondrial. Este ADN solo es registrado por vía matriarcal, Un nieto hereda el 100% del ADN mitocondrial con madre, abuela, las hijas de la abuela y sus primos en línea matriarcal. Para los hombres se encuentra el cromosoma Y que tampoco puede recombinar, lo cual permite analizar a un individuo contra su padre, abuelo, hijos masculinos del abuelo y primos varones. Por esta razón, en los análisis de identificación humana, dependiendo del objetivo, se emplean marcadores localizados ya sea en los cromosomas autosómicos “identificación de muestras biológicas”, cromosoma Y (paternidad) y ADN mitocondrial (maternidad). Para todos los casos se han identificado marcadores microsatélite a nivel forense.
Alelos repetidos al interior de una población
El problema con los marcadores genéticos es que no podemos emplearlos de cualquier forma, tomemos por ejemplo el siguiente caso. Asumimos una población mendeliana y analizamos un marcador con dos alelos posibles en una población cercana al equilibrio H-W. La madre posee el genotipo A1A2, el hijo “H” A2A2 y dos posibles padres, donde el supuesto padre 1 “P1” es A1A1 y el supuesto padre 2 “P2” es A2A2. En este caso podemos excluir fácilmente a P1 ya que no existe posibilidad de que él pueda ofrecer el alelo A2 del locus A, pero eso no implica que P2 sea el padre de H.
Si tomáramos una muestra al azar de la población donde se encuentra la mujer podríamos obtener una población relativamente grande, de cientos hasta miles de individuos que portan el alelo A2 y por lo tanto podrían ser implicados como posibles progenitores. Así que el problema recae en ¿Cómo excluir al mayor número de personas, con la menor cantidad de recursos?
La primera solución para responder al problema de las repeticiones es emplear un locus multivariable, es decir, donde la cantidad de alelos sea mayor a dos. Muchos alelos loci microsatélite pueden tener entre 5 y 20 alelos posibles, lo cual facilita los análisis.
Equilibrio Hardy-Weinberg
Este es quizá el factor más debatido entre la comunidad científica a la hora de determinar los factores que influenciarán los cálculos sobre la paternidad, en cualquier caso, explicaremos los fundamentos basados en un mundo ideal. En un mundo ideal se pueden cumplir las condiciones ideales del equilibrio Hardy-Weinberg, lo cual nos permite comparar una muestra de marcadores genéticos contra las probabilidades de haber acusado a un fulano al azar dentro de la población.
En otras palabras, el equilibrio H-W nos permitirá posteriormente determinar qué tan probable es que nuestra muestra pertenezca al individuo en cuestión, y no a un individuo al azar. En primera instancia, debe realizarse un muestreo ideal, donde se determina a una gran cantidad de individuos de la población al azar “en un mundo ideal, todos los individuos de la población deberían ser muestreados”, en un mundo ideal los individuos de la población no poseen subcategorías como raza, nacionalidad, etnicidad, estratificación económica, por lo que la reproducción es en términos prácticos, al azar.
Ahora digamos que el alelo A11 tiene una probabilidad de 0.0015. ¿Cuantos serán los posibles portadores en una población de 500 000 habitantes? ¿Cuántos serán los portadores en una población de 5 000 000 habitantes? El cálculo se realiza mediante la fórmula.

Donde Njo es el número de individuos que esperamos que tengan el alelo determinado j, Pjo es la probabilidad teórica del alelo j expresada como frecuencias, ya sean decimales o fracciones, y N es el número total de individuos en una población dada. Recuerde que la probabilidad es una frecuencia adimensional y que el número de individuos también es adimensional.
¿Cuántos serán los posibles portadores en una población de 500 000 habitantes y para otra de 5 000 000? Asuma que la probabilidad del alelo A11es de 0.0015
Solución analítica: aplicamos la ecuación 1.
Solución numérica: En una relación
No(A11) = 0.0015 × 500 000 = 750
Se espera que 750 individuos porten el alelo sin importar el sexo, para solo machos se esperaría la mitad, 375 hombres. Una población 10 veces más grande tendrá 3750 hombres portadores esperados.
En otras palabras, si nuestro caso ha ocurrido en una ciudad mediana de cinco millones, existe aproximadamente 3750 hombres que podrían ser acusados de ser el padre irresponsable.
Usando probabilidades anidadas
Existe una alternativa que puede limitar astronómicamente la cantidad de sospechosos, y es mediante el empleo de más de un loci en la prueba de paternidad. En nuestro caso, al adicionar un solo alelo multivariable más, podemos asegurar la exclusión de los posibles padres de nuestra muestra. Esto sucede debido a que al usar dos loci independientes, la probabilidad de una determinada combinación genética se obtiene por la multiplicación de las probabilidades individuales. Si usamos dos loci, la ecuación 1 se puede modificar de la siguiente manera.

En este caso el multiplicatorio es el producto de las probabilidades de varios genes, desde 1 hasta el último (z), además de la probabilidad de ser macho en una población completa. En una población grande hay que tener en cuenta el número de individuos y la posibilidad de flujo genético y mutaciones, por lo que los análisis se realizan generalmente con 5 o 6 loci con más de 10 alelos.
Índice de exclusión e inclusión
El índice de exclusión es un cálculo de probabilidad que determina que tan probable es encontrar a otros individuos al azar que posean el genotipo necesario para ser el progenitor, valor que se obtiene al multiplicar las probabilidades individuales.

Debido a la seriedad de esto, la probabilidad de cada alelo no es teórica, sino que es una medición poblacional. El índice de exclusión es la probabilidad de no encontrar individuos al azar con la combinación genética dada.

Debido a la seriedad de esto, la probabilidad de cada alelo no es teórica, sino que es una medición poblacional. El objetivo de los genetistas forenses es disminuir lo las posible Pinc para que los abogados no tengan la posibilidad de decir que existe una cantidad X en la población que también deberían estar investigados en un determinado caso.

Índice de paternidad
El índice de paternidad es otro estadístico que sirve para correlacionar los resultados obtenidos a través de la determinación de marcadores como los mini y microsatélites. La natación del índice de paternidad es la siguiente:

Donde Xi es la probabilidad de herencia mendeliana, así un homocigoto que posee los dos alelos concordantes tendrá un valor de 1, mientras que un heterocigoto tendrá un valor de 0,5, mientras que el homocigoto sin el alelo buscado tendrá un valor de cero. Este modelo estadístico indica que, con tan solo un locus con una combinación excluyente, el índice de paternidad será cero y el individuo será excluido de la paternidad. Para que sea útil, el índice de paternidad debe analizarse para varios loci:
Índice combinatorio de paternidad. Los valores del IC se transforman a probabilidad de parentesco mediante la siguiente fórmula:

Miremos el siguiente ejemplo.

Mutaciones
Uno de los presupuestos del equilibrio Hardy-Weinberg que puede resultar muy problemático es la ausencia de mutación, ya que en un sistema real como una población humana si hay mutaciones espontáneas. Es más, los microsatélites son útiles debido a que su tasa de mutación es bastante alta, por lo que pueden darse casos en que se podría generar una falsa exclusión debido a la mutación en un locus microsatélite. Es por esta razón que se emplean como mínimo 5 marcadores, si hay discrepancia en 1 pero los demás se mantienen puede expandirse el análisis a 10 marcadores microsatélite lo cual refuerza los cálculos estadísticos, es importante nunca confiarse de solo una línea de evidencias.
Representación de un marcador microsatélite y dilemas éticos
Los marcadores microsatélite generalmente están determinados por un código de gen-locus y por un código de alelo. El código de gen puede hacer referencia al cromosoma y lugar de ubicación u a otros criterios más específicos, mientras que el código de alelo hace referencia a la cantidad de repeticiones. Un ejemplo de esto puede verse en el siguiente cuadro. Debido a que existe loci microsatélite asociado a genes y a enfermedades, su análisis se encuentra prohibido en términos de identificación humana, debido a la carga legal que esto implica “en términos de seguros médicos”. Si le tenían miedo a un futuro donde los seguros pueden analizar tus enfermedades y tu potencial de desviaciones mentales, pues sorpresa, eso ya es el presente.
R. Referencias bibliográficas
R. Referencias bibliográficas
Bailey, J. A., Gu, Z., Clark, R. A., Reinert, K., Samonte, R. V, Schwartz, S., … Eichler, E. E. (2002). Recent segmental duplications in the human genome. Science, 297(5583), 1003–1007.
Cardoso-Moreira, M., & Long, M. (2012). The origin and evolution of new genes. En Evolutionary Genomics (pp. 161–186). Springer.
Carey, N. (2015). Junk DNA: a journey through the dark matter of the genome. Columbia University Press.
Carroll, S. B. (2008). Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell, 134(1), 25–36.
Conant, G. C., & Wolfe, K. H. (2008). Turning a hobby into a job: how duplicated genes find new functions. Nature Reviews Genetics, 9(12), 938.
Consortium, E. P. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature, 489(7414), 57.
Consortium, I. H. G. S. (2001). Initial sequencing and analysis of the human genome. nature, 409(6822), 860.
Cusanelli, E., & Chartrand, P. (2014). Telomeric noncoding RNA: telomeric repeat‐containing RNA in telomere biology. Wiley Interdisciplinary Reviews: RNA, 5(3), 407–419.
Harrison, P. M., Echols, N., & Gerstein, M. B. (2001). Digging for dead genes: an analysis of the characteristics of the pseudogene population in the Caenorhabditis elegans genome. Nucleic acids research, 29(3), 818–830.
Hawkins, J. S., Kim, H., Nason, J. D., Wing, R. A., & Wendel, J. F. (2006). Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome research, 16(10), 1252–1261.
Hayashi, Y., Aita, T., Toyota, H., Husimi, Y., Urabe, I., & Yomo, T. (2006). Experimental rugged fitness landscape in protein sequence space. PLoS One, 1(1), e96.
Hooper, J. E. (2014). A survey of software for genome-wide discovery of differential splicing in RNA-Seq data. Human genomics, 8(1), 3.
Karp, G. C. (2013). Cell and Molecular Biology, Concepts and Experiments (7a ed.). USA: Wiley Online Library.
Kellis, M., Wold, B., Snyder, M. P., Bernstein, B. E., Kundaje, A., Marinov, G. K., … Dekker, J. (2014). Defining functional DNA elements in the human genome. Proceedings of the National Academy of Sciences, 111(17), 6131–6138.
Klug, W. S., Cummings, M. R., Spencer, C. A., & Palladino, M. A. (2012). Concepts of Genetics (10a ed.). USA: Pearson.
Li, M., Marin-Muller, C., Bharadwaj, U., Chow, K.-H., Yao, Q., & Chen, C. (2009). MicroRNAs: control and loss of control in human physiology and disease. World journal of surgery, 33(4), 667–684.
Lieberman, M. A., & Rice, R. (2014). Biochemistry, Molecular, Biology, and Genetics (6a ed.). Lippincott Williams & Wilkins.
Lopez, J. V, Yuhki, N., Masuda, R., Modi, W., & O’Brien, S. J. (1994). Numt, a recent transfer and tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat. Journal of molecular evolution, 39(2), 174–190.
Maere, S., De Bodt, S., Raes, J., Casneuf, T., Van Montagu, M., Kuiper, M., & Van de Peer, Y. (2005). Modeling gene and genome duplications in eukaryotes. Proceedings of the National Academy of Sciences, 102(15), 5454–5459.
Maranda, V., Sunstrum, F. G., & Drouin, G. (2019). Both male and female gamete generating cells produce processed pseudogenes in the human genome. Gene, 684, 70–75.
Marshall, C. R., Raff, E. C., & Raff, R. A. (1994). Dollo’s law and the death and resurrection of genes. Proceedings of the National Academy of Sciences, 91(25), 12283–12287.
Morris, K. V. (2012). Non-coding RNAs and epigenetic regulation of gene expression: Drivers of natural selection. Horizon Scientific Press.
Nielsen, H., & Johansen, S. D. (2009). Group I introns: moving in new directions. RNA biology, 6(4), 375–383.
Ohno, S. (1970). Evolution by Gene Duplication Springer, New York.
Pennisi, E. (2012). ENCODE project writes eulogy for junk DNA. American Association for the Advancement of Science.
Petrov, D. A., & Hartl, D. L. (2000). Pseudogene evolution and natural selection for a compact genome. Journal of Heredity, 91(3), 221–227.
Qiu, Y., Taylor, A. B., & McMANUS, H. A. (2012). Evolution of the life cycle in land plants. Journal of Systematics and Evolution, 50(3), 171–194.
Rouquier, S., Blancher, A., & Giorgi, D. (2000). The olfactory receptor gene repertoire in primates and mouse: evidence for reduction of the functional fraction in primates. Proceedings of the National Academy of Sciences, 97(6), 2870–2874.
Tutar, Y. (2012). Pseudogenes. Comparative and functional genomics, 2012.
Visel, A., Rubin, E. M., & Pennacchio, L. A. (2009). Genomic views of distant-acting enhancers. Nature, 461(7261), 199.
Wapinski, I., Pfeffer, A., Friedman, N., & Regev, A. (2007). Natural history and evolutionary principles of gene duplication in fungi. Nature, 449(7158), 54.
William, A. D., & Ruse, M. (2004). Debating design: from Darwin to DNA.
Xie, J., Chen, S., Xu, W., Zhao, Y., & Zhang, D. (2019). Origination and Function of Plant Pseudogenes. Plant signaling & behavior, 14(8), 1625698.
Zhang, F., Zhou, Z., Xu, X., & Wang, X. (2002). A juvenile coelurosaurian theropod from China indicates arboreal habits. Naturwissenschaften, 89(9), 394–398.
Zhang, J. (2003). Evolution by gene duplication: an update. Trends in ecology & evolution, 18(6), 292–298.
Zhang, J., Zhang, Y., & Rosenberg, H. F. (2002). Adaptive evolution of a duplicated pancreatic ribonuclease gene in a leaf-eating monkey. Nature genetics, 30(4), 411.
Zheng, D., Frankish, A., Baertsch, R., Kapranov, P., Reymond, A., Choo, S. W., … Snyder, M. (2007). Pseudogenes in the ENCODE regions: consensus annotation, analysis of transcription, and evolution. Genome research, 17(6), 839–851.
No hay comentarios:
Publicar un comentario