
Índice
Índice
||1|| Introducción ||2|| Historia de la termoquímica ||3|| La energía ||4|| Sistema, alrededores y calor ||5|| La primera ley de la termodinámica ||6|| Entalpía ||7|| Entalpía de la reacción | ||10|| Calorimetría a presión constante ||11|| Ley de Hess ||12|| Entalpías de formación ||13|| Calor de solución y disolución ||14|| Alimentos ||15|| Combustibles fósiles y otras fuentes de energía ||16|| La energía del futuro |
Portada

Portada
1. Introducción
1. Introducción
Todo lo que hacemos está conectado de una forma u otra con la energía. No solo nuestra sociedad moderna sino la vida misma depende de la energía para su existencia. Los problemas relacionados con la energía (sus fuentes, producción, distribución y consumo) impregnan las conversaciones en ciencia, política y economía, y se relacionan con las preocupaciones ambientales y las políticas públicas.
Con la excepción de la energía del Sol, la mayor parte de la energía utilizada en nuestra vida diaria proviene de reacciones químicas. La combustión de gasolina, la producción de electricidad a partir de carbón, el calentamiento de viviendas por gas natural y el uso de baterías para alimentar dispositivos electrónicos son ejemplos de cómo se utiliza la química para producir energía. Incluso las células solares, dependen de la química para producir el silicio y otros materiales que convierten la energía solar directamente en electricidad. Además, las reacciones químicas proporcionan la energía que sustenta los sistemas vivos. Las plantas usan energía solar para llevar a cabo la fotosíntesis, lo que les permite crecer. Las plantas a su vez proporcionan alimentos de los cuales los humanos derivamos la energía necesaria para moverse, mantener la temperatura corporal y llevar a cabo todas las demás funciones corporales.
Es evidente que el tema de la energía está íntimamente relacionado con la química. Sin embargo, ¿qué es exactamente la energía y qué principios están involucrados en su producción, consumo y transformación de una forma a otra?
En este capítulo comenzamos a explorar la energía y sus cambios. Estamos motivados no solo por el impacto de la energía en tantos aspectos de nuestra vida diaria, sino también porque si queremos comprender adecuadamente la química, debemos comprender los cambios de energía que acompañan a las reacciones químicas.
El estudio de la energía y sus transformaciones se conoce como termodinámica (en griego: thérme-, "calor"; dy’namis, "poder"). Esta área de estudio comenzó durante la Revolución Industrial para desarrollar las relaciones entre el calor, el trabajo y los combustibles en las máquinas de vapor. En este capítulo examinaremos las relaciones entre las reacciones químicas y los cambios de energía que involucran calor. Esta parte de la termodinámica se llama termoquímica.
(1.1) Wilhelm Ostwald
Wilhelm Ostwald fue un físico y químico alemán, nacido en 1853. Es conocido por sus contribuciones a la termodinámica y la química coloidal, y por ser uno de los fundadores de la revista Zeitschrift für Physikalische Chemie. Fue galardonado con el Premio Nobel de Química en 1909 por sus estudios sobre la velocidad de las reacciones químicas y su aplicación a la termodinámica química. Además de su trabajo científico, también fue un defensor del internacionalismo y la paz mundial. Ostwald falleció en 1932.

Contexto social
Wilhelm Ostwald nació en Riga, entonces parte del Imperio Ruso, en 1853. Durante su vida, experimentó una época de cambios políticos y sociales significativos en Europa.
En cuanto al contexto político, durante su juventud, los territorios bálticos estaban bajo el dominio del Imperio Ruso, pero después de la Primera Guerra Mundial, Riga se convirtió en la capital de la nueva república de Letonia. Además, durante su vida, Ostwald fue testigo de importantes acontecimientos políticos, como el surgimiento del movimiento socialista y las tensiones políticas entre las grandes potencias europeas que eventualmente llevarían a la Primera Guerra Mundial.
En cuanto al contexto económico, Ostwald vivió durante una época de intensa industrialización y creciente globalización. Esto se refleja en su trabajo científico, en el que hizo importantes contribuciones a la química aplicada, especialmente en el campo de la termodinámica. También vivió en una época de intensos cambios sociales y culturales, en la que se produjeron grandes avances en la ciencia y la tecnología, así como en las artes y la cultura.
En cuanto al contexto religioso y cultural, Ostwald creció en un entorno luterano y estuvo fuertemente influenciado por la filosofía de Ernst Mach y el positivismo científico. Además, fue un defensor del internacionalismo y la paz mundial, y creía en la importancia de la cooperación científica internacional.
Infancia
Wilhelm Ostwald nació en Riga, Letonia, el 2 de septiembre de 1853. Era el mayor de siete hermanos y su familia pertenecía a la clase media. Su padre era un comerciante exitoso y la familia tenía una buena posición económica.
Desde joven, Ostwald mostró un gran interés por la ciencia y la naturaleza, y pasaba mucho tiempo explorando los campos y bosques cercanos a su hogar. Además, mostró una gran habilidad para el dibujo y la pintura, y sus padres le animaron a desarrollar estas habilidades.
Logros
Wilhelm Ostwald comenzó sus estudios básicos en una escuela privada luterana en Riga, Letonia, donde su familia residía. Allí recibió una educación general y religiosa, pero también mostró interés por la ciencia y la naturaleza desde una edad temprana.
Después de completar sus estudios de escuela secundaria, Ostwald se trasladó a Tartu, en Estonia, para comenzar sus estudios universitarios en la Universidad de Tartu en 1872. Allí estudió química, física y matemáticas, y recibió su doctorado en química en 1878 con una tesis sobre la teoría de los ácidos.
Después de completar su doctorado, Ostwald trabajó como profesor de química en la Universidad de Riga y en la Universidad Técnica de Dresde. Durante su carrera, hizo importantes contribuciones a la termodinámica y la química coloidal, y desarrolló nuevos métodos para medir la presión osmótica de las soluciones y para determinar el peso molecular de los polímeros. También fue uno de los fundadores de la revista Zeitschrift für Physikalische Chemie, que se convirtió en una de las publicaciones científicas más importantes en el campo de la química física.
Además de sus logros científicos, Ostwald también fue un defensor del internacionalismo y la paz mundial. Después de la Primera Guerra Mundial, se unió a la Sociedad de Naciones y fue nombrado presidente del Comité Internacional de la Cruz Roja. Por sus contribuciones científicas, Ostwald recibió numerosos premios y reconocimientos, incluyendo el Premio Nobel de Química en 1909.
Controversias
Wilhelm Ostwald fue parte de la comunidad científica de finales del siglo XIX y principios del XX, que experimentó grandes avances en la química y otras áreas de la ciencia. Entre sus principales colaboradores se encontraban los científicos físicos y químicos más destacados de la época, incluyendo a Svante Arrhenius, Friedrich Kohlrausch, y Jacobus Henricus van 't Hoff.
Uno de los mayores debates de la época en la que Ostwald trabajaba era la teoría atómica de la materia. Aunque esta teoría había sido propuesta desde la antigua Grecia, no fue hasta el siglo XIX cuando comenzó a ser aceptada por la comunidad científica. Sin embargo, algunos científicos, incluyendo a Ostwald, cuestionaron su validez.
Ostwald argumentó que los átomos y las moléculas no podían ser observados directamente, y que su existencia se basaba únicamente en inferencias. Creía que la química debería centrarse en los aspectos cuantitativos y medibles de las reacciones químicas, en lugar de en la teoría atómica.
Esto le llevó a enfrentarse a otros científicos de la época, como Dmitri Mendeléyev, quien desarrolló la tabla periódica de los elementos basándose en la teoría atómica. Sin embargo, aunque no estuvo de acuerdo con la teoría atómica, Ostwald siguió haciendo importantes contribuciones a la química, y en 1909 recibió el Premio Nobel de Química por sus investigaciones sobre la catálisis.
Aunque Wilhelm Ostwald nunca aceptó completamente la teoría atómica, su opinión sobre ella cambió con el tiempo gracias a las pruebas experimentales que se realizaron en la época.
Uno de los científicos que más influyó en la opinión de Ostwald sobre la teoría atómica fue el físico y químico alemán Max Planck. Planck defendió la teoría atómica y argumentó que era la única forma de explicar los fenómenos que se estaban observando en la física y la química.
Además, Planck desarrolló la teoría cuántica, que demostró que los átomos y las moléculas eran reales y no solo una construcción teórica. Este descubrimiento fue crucial para el desarrollo de la física y la química en el siglo XX.
Si bien Ostwald nunca aceptó completamente la teoría atómica, Planck logró convencerlo de que los átomos y las moléculas eran una parte fundamental de la comprensión de la química y la física. A pesar de que nunca aceptó completamente la teoría atómica, Ostwald reconoció su importancia en la explicación de los fenómenos químicos y físicos. De lo anterior se entiende que Ostwald acuñara el término “ficción útil”. En su libro "The Philosophy of Chemistry" (1891) Ostwald argumentó que los átomos y las moléculas eran conceptos teóricos que no podían ser observados directamente, y que su existencia se basaba únicamente en inferencias a partir de los resultados experimentales. Sin embargo, a pesar de que los átomos y las moléculas eran una construcción teórica, eran útiles para explicar y predecir los fenómenos químicos y físicos. Por lo tanto, la idea de que los átomos son una "ficción útil" se refiere a la idea de que, aunque no podemos observar directamente los átomos, son una herramienta útil para entender y explicar la naturaleza de la materia.
Logros
Wilhelm Ostwald recibió muchos reconocimientos y honores en su vida y después de su muerte. En 1909, recibió el Premio Nobel de Química por sus trabajos sobre la catálisis, la equilibrio químico y la termodinámica. Además del Premio Nobel, Ostwald también recibió numerosos títulos honoríficos y medallas. Fue elegido miembro de muchas sociedades científicas, incluyendo la Real Sociedad de Londres, la Academia de Ciencias de Francia y la Academia Nacional de Ciencias de Estados Unidos. En 1915, fue nombrado barón por el rey de Sajonia y recibió el título de "Wilhelm von Ostwald". También fue elegido presidente de la Asociación Alemana de Químicos en 1903 y fue galardonado con la Medalla Davy por la Royal Society de Londres en 1922.
Después de su muerte en 1932, muchos homenajes y reconocimientos fueron realizados en su honor. En 1934, se creó el Premio Wilhelm Ostwald, que se otorga anualmente por la Sociedad Alemana de Química. Además, la Universidad de Leipzig, donde Ostwald fue profesor durante muchos años, estableció una cátedra en su honor. También se erigieron monumentos y placas conmemorativas en su ciudad natal de Riga y en Leipzig. En resumen, Wilhelm Ostwald fue un científico muy reconocido en su época y recibió numerosos honores y premios por sus contribuciones a la química y la ciencia en general.
Importancia
El trabajo de Wilhelm Ostwald ha tenido un impacto significativo en la vida cotidiana de las personas, especialmente en el campo de la química. Algunas de sus contribuciones más importantes incluyen:
(a) Desarrollo de la teoría de la catálisis: La teoría de Ostwald sobre la catálisis ha sido fundamental para el desarrollo de procesos industriales que involucran reacciones químicas. La catálisis es utilizada en la producción de una amplia variedad de productos, como plásticos, medicamentos, productos químicos y combustibles.
(b) Establecimiento de la termodinámica química: Ostwald fue uno de los primeros científicos en desarrollar la termodinámica química, que ha permitido a los químicos comprender mejor cómo los cambios de temperatura y presión afectan a las reacciones químicas. Esta comprensión ha llevado al desarrollo de procesos industriales más eficientes y sostenibles.
(c) Desarrollo de la teoría ácido-base: Ostwald desarrolló la teoría ácido-base, que ha sido fundamental para la comprensión de cómo los ácidos y bases interactúan en solución. Esta teoría es importante en muchos campos, incluyendo la química, la biología y la medicina.
¿Qué debemos aprender de el?
La vida de Wilhelm Ostwald es una fuente de inspiración para cualquiera que aspire a alcanzar grandes logros y dejar un impacto duradero en el mundo. A lo largo de su carrera, Ostwald enfrentó muchos obstáculos y limitaciones, pero siempre perseveró y trabajó arduamente para superarlos.
Una de las lecciones más importantes que podemos aprender de la vida de Ostwald es la importancia de la curiosidad intelectual y el pensamiento crítico. Ostwald siempre estuvo dispuesto a cuestionar las teorías establecidas y buscar nuevas formas de entender el mundo. Además, nunca se dio por vencido ante los obstáculos, sino que se mantuvo firme en su compromiso de alcanzar sus objetivos.
Otra lección que podemos aprender de la vida de Ostwald es la importancia de trabajar en colaboración con otros. Ostwald tuvo muchos colaboradores a lo largo de su carrera y siempre estuvo dispuesto a compartir sus ideas y conocimientos con otros científicos. Este enfoque colaborativo lo ayudó a alcanzar muchos de sus logros más importantes.
Por último, la vida de Ostwald nos recuerda la importancia de dejar un impacto duradero en el mundo. A través de su trabajo en la química, Ostwald ha dejado un legado que continúa influyendo en nuestras vidas cotidianas hoy en día. Su enfoque en la sostenibilidad y la eficiencia también es un recordatorio de la importancia de trabajar por el bien común y el futuro del planeta.
En resumen, la vida de Wilhelm Ostwald es una fuente de inspiración y nos recuerda la importancia de la curiosidad intelectual, el pensamiento crítico, la colaboración y dejar un impacto duradero en el mundo.
2. Historia de la termoquímica
2. Historia de la termoquímica
La historia de la termodinámica es un hilo fundamental en la historia de la física, la historia de la química y la historia de la ciencia en general. Debido a la relevancia de la termodinámica en gran parte de la ciencia y la tecnología, su historia está finamente entretejida con los desarrollos de la mecánica clásica, la mecánica cuántica, el magnetismo y la cinética química, hasta campos aplicados más lejanos como la meteorología, la teoría de la información y la biología, y a desarrollos tecnológicos como la máquina de vapor, el motor de combustión interna, la criogenia y la generación de electricidad. El desarrollo de la termodinámica impulsó y fue impulsado por la teoría atómica.
(2.1) De la antigüedad a la edad media
Los antiguos veían el calor como algo relacionado con el fuego. En el año 3000 a. C., los antiguos egipcios consideraban que el calor estaba relacionado con las mitologías del origen (Griffiths, 1955). En la tradición filosófica occidental, después de mucho debate sobre el elemento primordial entre los primeros filósofos presocráticos, Empédocles propuso una teoría de los cuatro elementos, en la que todas las sustancias provienen de la tierra, el agua, el aire y el fuego. El elemento empedocleano del fuego es quizás el principal antepasado de conceptos posteriores como el flogisto y el calórico. Alrededor del año 500 a. C., el filósofo griego Heráclito se hizo famoso como el filósofo del "flujo y el fuego" por su frase proverbial: "Todas las cosas fluyen". Heráclito argumentó que los tres elementos principales de la naturaleza eran el fuego, la tierra y el agua (Siddiqui, 2018).
El atomismo es una parte central de la relación actual entre la termodinámica y la mecánica estadística. Pensadores antiguos como Leucipo y Demócrito, y más tarde los epicúreos, al promover el atomismo, sentaron las bases para la teoría atómica posterior. Hasta que más tarde se proporcionaron pruebas experimentales de los átomos en el siglo XX, la teoría atómica fue impulsada en gran medida por consideraciones filosóficas e intuición científica. En consecuencia, los filósofos antiguos utilizaron la teoría atómica para llegar a conclusiones que hoy pueden considerarse inmaduras o ingenuas: por ejemplo, Demócrito da una vaga descripción atomista del alma, a saber, que está "construida a partir de átomos delgados, lisos y redondos, similares a los de fuego" (Cercignani, 2001; Chalmers, 2009; Onorato, Malgieri, Polesello, Salmoiraghi, & Oss, 2019).
En el siglo V a. C., el filósofo griego Parménides, en su única obra conocida, un poema convencionalmente titulado Sobre la naturaleza, usa el razonamiento verbal para postular que un vacío, esencialmente lo que ahora se conoce como vacío, en la naturaleza no podría ocurrir. Este punto de vista fue apoyado por los argumentos de Aristóteles, pero fue criticado por Leucipo y Heron de Alejandría. Desde la antigüedad hasta la Edad Media, se presentaron varios argumentos para probar o desaprobar la existencia de un vacío y se hicieron varios intentos para construir un vacío, pero todos fracasaron (Kenny, 1964; Redhead, 1999).

Figura 2.1. Santorio Santorio (1561-1636) fue un médico italiano nacido en Capodistria (actualmente Koper, Eslovenia). Estudió medicina en Padua y fue discípulo de Girolamo Mercuriale. Santorio es conocido por sus estudios en fisiología, especialmente por la invención del primer termómetro médico. También fue uno de los primeros en utilizar la balanza para medir la pérdida de peso en pacientes. Sus investigaciones sobre la nutrición y la digestión también son destacables. Santorio es considerado un pionero en el estudio de la medicina experimental y su obra "Ars de statica medicina" es un clásico de la medicina moderna.
Los científicos europeos Cornelius Drebbel, Robert Fludd, Galileo Galilei y Santorio Santorio en los siglos XVI y XVII pudieron medir la relativa "frialdad" o "calor" del aire, utilizando un termómetro de aire rudimentario (o termoscopio). Esto puede haber sido influenciado por un dispositivo anterior que podía expandir y contraer el aire construido por Filón de Bizancio y Heron de Alejandría (Buyse, 2020; Taylor, 1942).
Alrededor de 1600, el filósofo y científico inglés Francis Bacon conjeturó: "El calor en sí mismo, su esencia y esencia es movimiento y nada más". En 1643, Galileo Galilei, aunque generalmente aceptaba la explicación de "succión" del horror vacui, es decir que el vacío no puede existir porque inmediatamente succiona algo a su alrededor y se llena, propuesta por Aristóteles, también creía que esta succión por parte del vacío era limitada. Las bombas que operan en las minas ya habían demostrado que la naturaleza solo llenaría un vacío con agua hasta una altura de ~ 30 pies. Conociendo este curioso dato, Galileo animó a su exalumno Evangelista Torricelli a investigar estas supuestas limitaciones. Torricelli no creía que el (Horror vacui/poder de succión del vacío), en el sentido de la perspectiva de "succión" de Aristóteles, fuera responsable de hacer subir el agua. Más bien, razonó, era el resultado de la presión ejercida sobre el líquido por el aire circundante (Middleton, 1963; West, 2013).

Figura 2.2. Evangelista Torricelli (1608-1647) fue un físico y matemático italiano nacido en Faenza. Estudió matemáticas y filosofía en Roma y posteriormente trabajó como asistente de Galileo Galilei. Torricelli es famoso por su invención del primer barómetro, que permitió medir la presión atmosférica y sentó las bases para el estudio de la meteorología. También hizo importantes contribuciones a la geometría y la trigonometría, y es conocido por la "fórmula de Torricelli" para el cálculo del volumen de un sólido de revolución. Además, Torricelli es considerado uno de los fundadores del cálculo integral junto con su contemporáneo Blaise Pascal. Torricelli murió a la edad de 39 años, pero sus contribuciones a la física y las matemáticas siguen siendo recordadas y valoradas en la actualidad.
Para probar esta hipótesis, llenó un tubo largo de vidrio (sellado en un extremo) con mercurio y lo volcó en un plato que también contenía mercurio. Solo se vació una parte del tubo; Quedaban ~30 pulgadas del líquido. A medida que el mercurio se vaciaba, se creaba un vacío en la parte superior del tubo. Este, el primer vacío hecho por el hombre, refutó efectivamente la teoría de la "succión" de Aristóteles y afirmó la existencia de vacíos en la naturaleza. La fuerza gravitacional sobre el elemento pesado que es Mercurio le impidió llenar el vacío. La naturaleza puede aborrecer el vacío, pero a la gravedad no le importa (Middleton, 1963; West, 2013).
(2.2) Transición de la química a la termoquímica
La teoría del flogisto surgió en el siglo XVII, al final del período de la alquimia. Su sustitución por la teoría calórica en el siglo XVIII es uno de los hitos históricos de la transición de la alquimia a la química. El flogisto era una sustancia hipotética que se suponía que se liberaba de las sustancias combustibles durante la combustión y de los metales durante el proceso de oxidación. También se suponía que el calórico, como el flogisto, era la "sustancia" de calor que fluiría de un cuerpo más caliente a un cuerpo más frío, calentándolo así.
Los primeros desafíos experimentales sustanciales a la teoría calórica surgieron en el trabajo de Rumford de 1798, cuando demostró que los cañones de hierro fundido perforados producían grandes cantidades de calor que atribuyó a la fricción, y su trabajo fue uno de los primeros en socavar la teoría calórica (Fox, 2006). El desarrollo de la máquina de vapor también centró la atención en la calorimetría y la cantidad de calor producido por diferentes tipos de carbón. La primera investigación cuantitativa sobre los cambios de calor durante las reacciones químicas fue iniciada por Lavoisier usando un calorímetro de hielo siguiendo la investigación de Joseph Black sobre el calor latente del agua (Lecoustre, 2009).
Más estudios cuantitativos realizados por James Prescott Joule a partir de 1843 proporcionaron fenómenos sólidamente reproducibles y ayudaron a colocar el tema de la termodinámica sobre una base sólida. William Thomson, por ejemplo, todavía intentaba explicar las observaciones de Joule dentro de un marco calórico hasta 1850. Sin embargo, la utilidad y el poder explicativo de la teoría cinética pronto comenzaron a desplazar al calórico y quedó obsoleta en gran medida a fines del siglo XIX. Joseph Black y Lavoisier hicieron contribuciones importantes en la medición precisa de los cambios de calor usando el calorímetro, un tema que se conoció como termoquímica (A. Brown, 1999; Newburgh & Leff, 2011).
(2.3) La calorimetría
Robert Hooke pensó que el calor es una propiedad del cuerpo que surge del movimiento o agitación de sus partes. De acuerdo con la teoría principal en ese momento, se pensaba que el calor consistía en un fluido autorepelente llamado "calórico", que también era un material similar al gas sin peso. En 1799, Sir Humphrey Davy, un químico y físico inglés, frotó dos trozos de hielo en el vacío y notó que el hielo se estaba derritiendo. El propósito era ver si podía generar calor por fricción, una idea contraria a la teoría calórica sostenida anteriormente. De acuerdo con la teoría calórica, el hielo se derretiría solo si se pusiera en contacto con un cuerpo más caliente, liberando un flujo de partículas calóricas en el hielo, haciendo que se derrita. En el siglo XVIII una serie de experimentos cuantitativos comenzaron a madurar las ideas sobre la naturaleza del calor (Meschel, 2020).

Figura 2.3. Joseph Black (1728-1799) fue un químico y físico escocés nacido en Bordeaux, Francia. Estudió medicina en la Universidad de Glasgow y posteriormente se trasladó a Edimburgo, donde fue profesor de química durante más de 20 años. Black es conocido por sus estudios sobre los gases, en los que descubrió la existencia del dióxido de carbono y desarrolló la teoría del calor latente. También hizo importantes contribuciones a la química, descubriendo y estudiando el magnesio y el bario, y desarrollando el concepto de neutralización química. Black es considerado uno de los precursores de la Revolución Química del siglo XVIII y sus contribuciones sentaron las bases de la química moderna.
La medición del calor tiene aproximadamente trescientos años de colorida historia. El primer colaborador conocido de esta historia es Joseph Black, médico y químico escocés. En 1761, dedujo con medidas precisas que la adición de calor al hielo en su punto de fusión o al agua en su punto de ebullición no da como resultado un cambio de temperatura. Sus observaciones del calor latente y la definición de calor específico señalaron el nacimiento de la termodinámica. Black fue el primer científico en distinguir entre temperatura y calor (Meschel, 2020).
El próximo hito en esta historia es el trabajo del primer químico que realizó experimentos cuantitativos con calor. En 1789, Antoine Lavoisier en colaboración con el matemático Pierre Simon de La Place construyó el primer calorímetro. Lavoisier estaba interesado en medir el calor involucrado en el proceso de respiración de un conejillo de Indias. Colocó al animal en un compartimento central cerrado rodeado de hielo. En 10 h, el animal emitió suficiente calor para derretir 13 oz de hielo, pero la temperatura de su cuerpo no cambió. Al comparar cuantitativamente la cantidad de aire caliente exhalado por el animal con la misma cantidad de aire caliente producido por la combustión del carbón, concluyó que el proceso de respiración era una reacción de combustión (Meschel, 2020).
El siguiente hito en el desarrollo de la termodinámica nos lleva al concepto de calor como forma de energía. El experimentalista precursor fue el leal y espía británico, que luchó contra la Guerra Revolucionaria Estadounidense, Sir Benjamin Thompson. Mientras trabajaba para el duque de Baviera (Graf Rumford), diseñó un nuevo proceso de perforación de cañones. Thompson observó que el proceso de perforación del cañón bajo el agua creaba cambios de temperatura medibles. Si bien sus resultados numéricos fueron toscos, estableció que el calor es una forma de energía en la década de 1790 (Meschel, 2020).
(2.4) Nacimiento de la termodinámica como ciencia moderna.
En sus orígenes, la termodinámica fue el estudio de los motores. Un precursor del motor fue diseñado por el científico alemán Otto von Guericke quien, en 1650, diseñó y construyó la primera bomba de vacío del mundo y creó el primer vacío del mundo conocido como los hemisferios de Magdeburg. Se vio obligado a hacer un vacío para refutar la suposición de Aristóteles de que "la naturaleza aborrece el vacío".

Figura 2.4. Otto von Guericke (1602-1686) fue un científico, inventor y político alemán nacido en Magdeburgo. Estudió leyes y filosofía en diferentes universidades y posteriormente se convirtió en alcalde de Magdeburgo. Guericke es conocido por sus estudios en física, especialmente por sus experimentos con vacío y electricidad estática. En 1654, inventó la primera máquina de vacío y utilizó una versión mejorada para realizar la famosa "Experiencia de los hemisferios de Magdeburgo", donde demostró la fuerza del vacío al separar dos hemisferios de cobre que habían sido unidos al vacío. También es reconocido por sus contribuciones al desarrollo de la bomba de aire, el barómetro y el electróforo. Guericke fue un hombre versátil que también se destacó en política, literatura y religión. Sus contribuciones a la ciencia y la tecnología lo convierten en uno de los científicos más importantes del siglo XVII.
Poco después, el físico y químico irlandés Robert Boyle se enteró de los diseños de Guericke y en 1656, en coordinación con el científico inglés Robert Hooke, construyó una bomba de aire. Usando esta bomba, Boyle y Hooke notaron la correlación de presión-volumen: P.V=constante. En ese momento, se suponía que el aire era un sistema de partículas inmóviles y no se interpretaba como un sistema de moléculas en movimiento. El concepto de movimiento térmico llegó dos siglos después. Por eso la publicación de Boyle en 1660 habla de un concepto mecánico: el resorte neumático (Boyle, 1911).
Más tarde, tras la invención del termómetro, se pudo cuantificar la propiedad temperatura. Esta herramienta le dio a Gay-Lussac la oportunidad de derivar su ley, que condujo poco después a la ley de los gases ideales. Pero, ya antes del establecimiento de la ley de los gases ideales, un socio de Boyle llamado Denis Papin construyó en 1679 un digestor de huesos, que es un recipiente cerrado con una tapa bien ajustada que confina el vapor hasta que se genera una alta presión.

Figura 2.5. Nicolas Léonard Sadi Carnot (1796-1832) fue un físico y matemático francés, considerado el padre de la termodinámica. Estudió en la École Polytechnique y en la École des Ponts et Chaussées, donde se especializó en ingeniería. En 1824, publicó su obra fundamental, "Reflexiones sobre la Potencia Motriz del Fuego", en la que estableció las bases de la teoría de la termodinámica y desarrolló el concepto de ciclo termodinámico. Este trabajo fue clave para la comprensión de la conversión de la energía térmica en trabajo mecánico, y sentó las bases para la Revolución Industrial. Además de sus contribuciones en termodinámica, Carnot también trabajó en hidráulica y en el desarrollo de la geometría analítica. Carnot murió a los 36 años, pero su trabajo continuó influyendo en el desarrollo de la física y la ingeniería durante décadas, y su legado es reconocido como uno de los más importantes en la historia de la ciencia.
Los diseños posteriores implementaron una válvula de liberación de vapor para evitar que la máquina explotara. Al observar cómo la válvula se movía rítmicamente hacia arriba y hacia abajo, Papin concibió la idea de un motor de pistón y cilindro. Sin embargo, no siguió adelante con su diseño. Sin embargo, en 1697, basándose en los diseños de Papin, el ingeniero Thomas Savery construyó el primer motor. Aunque estos primeros motores eran toscos e ineficientes, atrajeron la atención de los principales científicos de la época. Uno de esos científicos fue Sadi Carnot, el “padre de la termodinámica”, quien en 1824 publicó “Reflexiones sobre la fuerza motriz del fuego” (Carnot, 1824), un discurso sobre el calor, la potencia y la eficiencia del motor. Esto marca el comienzo de la termodinámica como ciencia moderna. Una máquina de vapor Watt, la máquina de vapor que impulsó la Revolución Industrial en Gran Bretaña y el mundo.
Desde entonces, antes de 1698 y de la invención de la máquina Savery, los caballos se usaban para accionar poleas unidas a baldes que sacaban agua de las minas de sal inundadas en Inglaterra. En los años siguientes, se construyeron más variaciones de máquinas de vapor, como la Newcomen Engine y, más tarde, la Watt Engine. Con el tiempo, estos primeros motores eventualmente se utilizarían en lugar de los caballos. Por lo tanto, cada motor comenzó a asociarse con una cierta cantidad de "caballos de fuerza" según la cantidad de caballos que había reemplazado. El principal problema de estos primeros motores era que eran lentos y torpes, convirtiendo menos del 2% del combustible de entrada en trabajo útil. En otras palabras, se tenían que quemar grandes cantidades de carbón (o madera) para producir solo una pequeña fracción de la producción de trabajo. De ahí nació la necesidad de una nueva ciencia de la dinámica del motor.
La mayoría cita el artículo de Sadi Carnot de 1824 Reflections on the Motive Power of Fire como el punto de partida de la termodinámica como ciencia moderna. Carnot definió "fuerza motriz" como la expresión del efecto útil que un motor es capaz de producir. Aquí, Carnot nos presentó la primera definición moderna de "trabajo": peso levantado a través de una altura. El deseo de comprender, a través de la formulación, este efecto útil en relación con el "trabajo" está en el centro de toda la termodinámica moderna.

Figura 2.6. James Prescott Joule (Salford, Reino Unido, 24 de diciembre de 1818-11 de octubre de 1889) fue un físico inglés, uno de los más notables físicos de su época, conocido sobre todo por sus investigaciones en termodinámica. Descubrió su relación con el trabajo mecánico, lo cual le condujo a la teoría de la energía. La unidad internacional de energía, calor y trabajo, el joule, fue bautizada en su honor. Trabajó con lord Kelvin para desarrollar la escala absoluta de la temperatura, hizo observaciones sobre la teoría termodinámica (efecto Joule-Thomson) y encontró una relación entre la corriente eléctrica que atraviesa una resistencia y el calor disipado, llamada actualmente ley de Joule. Después de numerosos experimentos, obtuvo el valor numérico del equivalente mecánico del calor. Contribuyó a explicar la teoría cinética de los gases. Fue «posiblemente el último autodidacta que hizo una contribución significativa al progreso de la ciencia».
En 1843, James Joule encontró experimentalmente el equivalente mecánico del calor. En 1845, Joule informó sobre su experimento más conocido, que involucraba el uso de un peso que caía para hacer girar una rueda de paletas en un barril de agua, lo que le permitió estimar un equivalente mecánico de calor de 819 ft·lbf/Btu (Joule, 1854). Esto condujo a la teoría de la conservación de la energía y explicó por qué el calor puede realizar un trabajo. El nombre "termodinámica", sin embargo, no llegó hasta unos veinticinco años más tarde cuando, en 1849, el matemático y físico británico William Thomson (Lord Kelvin) acuñó el término termodinámica en un artículo sobre la eficiencia de las máquinas de vapor (Newburgh & Leff, 2011).
En 1850, el famoso físico matemático Rudolf Clausius originó y definió el término entalpía H como el contenido total de calor del sistema, derivado de la palabra griega enthalpein que significa calentar, y definió el término entropía S como el calor perdido o convertido, desperdicio, derivado de la palabra griega entrepein que significa convertir (Clausius, 1857a).
En asociación con Clausius, en 1871, un matemático y físico escocés, James Clerk Maxwell, formuló una nueva rama de la termodinámica llamada Termodinámica estadística, que funciona para analizar un gran número de partículas en equilibrio, es decir, sistemas en los que no se producen cambios, de modo que solo sus las propiedades promedio como la temperatura T, la presión P y el volumen V se vuelven importantes.
Poco después, en 1875, el físico austriaco Ludwig Boltzmann formuló una conexión precisa entre la entropía S y el movimiento molecular: S = kBlogW., definiéndose en términos del número de estados posibles [W] que dicho movimiento podría ocupar, donde kB es la constante de Boltzmann ( que es igual a la constante de los gases ideales dividida entre el número de Avogadro). El año siguiente, 1876, fue un punto seminal en el desarrollo del pensamiento humano. Durante este período esencial, el ingeniero químico Willard Gibbs, la primera persona en América en obtener un doctorado en ingeniería (Yale), publicó un oscuro artículo de 300 páginas titulado: Sobre el equilibrio de las sustancias heterogéneas (Gibbs, 1878), en el que formuló una gran igualdad, la Ecuación de energía libre de Gibbs, que da una medida de la cantidad de "trabajo útil" alcanzable en los sistemas que reaccionan.

Figura 2.7. Josiah Willard Gibbs (1839-1903) fue un físico y químico estadounidense conocido como el "padre de la termodinámica química". Nació en New Haven, Connecticut, y estudió en la Universidad de Yale, donde más tarde enseñó durante más de tres décadas. Gibbs desarrolló la teoría de la termodinámica química, que permitió explicar el comportamiento de las sustancias químicas y sus reacciones. También realizó importantes contribuciones en el campo de la mecánica estadística y la termodinámica de las fases, estableciendo la base matemática para el estudio de las propiedades de la materia en diferentes estados físicos. Entre sus logros más destacados se encuentran el desarrollo de la regla de las fases de Gibbs, que describe el número de variables necesarias para definir el equilibrio en un sistema, y la formulación de la ley de la energía libre de Gibbs, que es fundamental para la comprensión de las reacciones químicas y los procesos termodinámicos en general. Willard Gibbs es reconocido como uno de los científicos más importantes de la historia de la física y la química, y su trabajo ha tenido una gran influencia en la ciencia moderna.
Sobre la base de estos cimientos, personas como Lars Onsager, Erwin Schrödinger e Ilya Prigogine, entre otros, funcionaron para llevar estos "conceptos" de motores a la vía pública de casi todas las ramas modernas de la ciencia.
(2.5) Teoría cinética
La idea de que el calor es una forma de movimiento es quizás antigua y ciertamente fue discutida por Francis Bacon en 1620 en su Novum Organum (Bacon, 1878). La primera reflexión científica escrita sobre la naturaleza microscópica del calor se encuentra probablemente en un trabajo de Mikhail Lomonosov, en el que escribió:
“(..) no se debe negar el movimiento por el hecho de que no se ve. ¿Quién negaría que las hojas de los árboles se mueven cuando son susurradas por el viento, a pesar de que no se puede observar desde grandes distancias? Así como en este caso el movimiento permanece oculto debido a la perspectiva, permanece oculto en cuerpos cálidos debido a los tamaños extremadamente pequeños de las partículas en movimiento. En ambos casos, el ángulo de visión es tan pequeño que no se puede ver ni el objeto ni su movimiento".
Durante los mismos años, Daniel Bernoulli publicó su libro Hidrodinámica (Bernoulli, 1738), en el que derivó una ecuación para la presión de un gas considerando las colisiones de sus átomos con las paredes de un recipiente. Demuestra que esta presión es dos tercios de la energía cinética promedio del gas en una unidad de volumen. Las ideas de Bernoulli, sin embargo, tuvieron poco impacto en la cultura calórica dominante. Bernoulli hizo una conexión con el principio vis viva de Gottfried Leibniz, una formulación temprana del principio de conservación de la energía, y las dos teorías se entrelazaron íntimamente a lo largo de su historia. Aunque Benjamin Thompson sugirió que el calor era una forma de movimiento como resultado de sus experimentos en 1798, no se intentó reconciliar los enfoques teóricos y experimentales, y es poco probable que estuviera pensando en el principio de vis viva.
Más tarde, John Herapath formuló de forma independiente una teoría cinética en 1820, pero asoció erróneamente la temperatura con el impulso en lugar de vis viva o energía cinética. Su trabajo finalmente falló la revisión por pares y fue descuidado. John James Waterston en 1843 proporcionó un relato en gran parte preciso, nuevamente de forma independiente, pero su trabajo recibió la misma recepción, fallando la revisión por pares.
El progreso adicional en la teoría cinética comenzó solo a mediados del siglo XIX, con los trabajos de Rudolf Clausius, James Clerk Maxwell y Ludwig Boltzmann. En su obra de 1857 Sobre la naturaleza del movimiento llamado calor (Clausius, 1857b), Clausius establece claramente por primera vez que el calor es la energía cinética promedio de las moléculas. Esto interesó a Maxwell, quien en 1859 derivó la distribución de cantidad de movimiento que más tarde lleva su nombre. Posteriormente, Boltzmann generalizó su distribución para el caso de gases en campos externos.
Boltzmann es quizás el contribuyente más importante a la teoría cinética, ya que introdujo muchos de los conceptos fundamentales de la teoría. Además de la distribución de Maxwell-Boltzmann mencionada anteriormente, también asoció la energía cinética de las partículas con sus grados de libertad. La ecuación de Boltzmann para la función de distribución de un gas en estados de no equilibrio sigue siendo la ecuación más efectiva para estudiar los fenómenos de transporte en gases y metales. Al introducir el concepto de probabilidad termodinámica como el número de microestados correspondientes al macroestado actual, demostró que su logaritmo es proporcional a la entropía.
(2.6) La entropía y la segunda ley
Aunque estaba trabajando con la teoría calórica, Sadi Carnot en 1824 sugirió que parte del calórico disponible para generar trabajo útil se pierde en cualquier proceso real. En marzo de 1851, mientras luchaba por llegar a un acuerdo con el trabajo de James Prescott Joule, Lord Kelvin comenzó a especular que había una pérdida inevitable de calor útil en todos los procesos. La idea fue enmarcada aún más dramáticamente por Hermann von Helmholtz en 1854, dando nacimiento al espectro de la muerte térmica del universo (Styer, 2019).

Figura 2.8. Hermann von Helmholtz (1821-1894) fue un físico y fisiólogo alemán que hizo importantes contribuciones en una variedad de campos científicos. Nació en la ciudad de Potsdam, Prusia (actual Alemania), y estudió medicina en la Universidad de Berlín. A lo largo de su carrera, realizó investigaciones en áreas como la termodinámica, la electricidad, la óptica, la fisiología y la psicología. Entre sus contribuciones más destacadas se encuentran la formulación de la ley de la conservación de la energía (también conocida como la primera ley de la termodinámica), la invención del oftalmoscopio (un instrumento utilizado para examinar el interior del ojo), la explicación de la percepción del color y la realización de experimentos para demostrar la velocidad de la transmisión nerviosa. Helmholtz también fue un líder en la educación científica y en la organización de la investigación en Alemania. Fue el primer director del Instituto de Física de la Universidad de Berlín y más tarde ocupó el cargo de Ministro de Educación en Prusia. Hermann von Helmholtz es reconocido como uno de los científicos más importantes del siglo XIX y su trabajo ha tenido una gran influencia en la física, la fisiología y la psicología modernas.
En 1854, William John Macquorn Rankine comenzó a utilizar en el cálculo lo que llamó su función termodinámica. Posteriormente se ha demostrado que esto es idéntico al concepto de entropía formulado por Rudolf Clausius en 1865. Clausius utilizó el concepto para desarrollar su declaración clásica de la segunda ley de la termodinámica el mismo año (Xue & Guo, 2019).
(2.7) Criogenia
En 1702 Guillaume Amontons introdujo el concepto de cero absoluto basado en observaciones de gases. En 1810, Sir John Leslie congeló agua en hielo artificialmente. La idea del cero absoluto fue generalizada en 1848 por Lord Kelvin. En 1906, Walther Nernst estableció la tercera ley de la termodinámica (Mendelssohn, 1977).
(2.8) Termodinámica química
La termodinámica química es el estudio de la interrelación de la energía con las reacciones químicas o con un cambio físico de estado dentro de los límites de las leyes de la termodinámica. Durante los años 1873-1876, el físico matemático estadounidense Josiah Willard Gibbs publicó una serie de tres artículos, el más famoso de los cuales fue On the Equilibrium of Heterogeneous Substances (Gibbs, 1878), en el que mostró cómo los procesos termodinámicos podían analizarse gráficamente estudiando la energía, la entropía, el volumen, la temperatura y la presión del sistema termodinámico de tal manera que se pueda determinar si un proceso ocurriría espontáneamente. A principios del siglo XX, químicos como Gilbert N. Lewis, Merle Randall y E. A. Guggenheim comenzaron a aplicar los métodos matemáticos de Gibbs al análisis de procesos químicos.
3. La energía
3. La energía
A diferencia de la materia, la energía no tiene masa y no puede mantenerse en nuestras manos “de hecho si alguien pudiera manifestar su ki “a la Dragon Ball” lo que aparecería en las manos no es energía, sino plasma, una forma de materia gaseosa ionizada con alta energía”.
A pesar de estos dilemas conceptuales, los efectos derivados de los intercambios energéticos pueden observarse y medirse. La energía es la capacidad de hacer trabajo o transferir calor. Antes de que podamos hacer uso de esta definición, debemos comprender los conceptos de trabajo y calor.
(3.1) Mecanismos de transferencia de energía
Un mecanismo de transferencia de energía es un proceso mediante el cual la energía se mueve de un lugar a otro o se convierte de una forma a otra. Es importante tener en cuenta que la energía en sí misma es una propiedad abstracta que no puede ser creada ni destruida, pero puede ser transferida y transformada. Algunos de los mecanismos de transferencia de energía más comunes son:
(a) Conducción: es la transferencia de energía térmica a través de un material sólido debido a una diferencia de temperatura. Esta transferencia de energía se produce cuando las partículas de un material vibran y transfieren esa vibración a las partículas adyacentes.
(b) Convección: es la transferencia de energía térmica a través del movimiento de un fluido. Esto puede ser natural, como en el caso de la circulación de aire caliente, o forzado, como en el caso de un ventilador.
(c) Radiación: es la transferencia de energía a través de ondas electromagnéticas, como la luz visible y la radiación infrarroja. Esto ocurre cuando un cuerpo emite energía en forma de ondas electromagnéticas que son absorbidas por otro cuerpo.
(d) Trabajo mecánico: es la transferencia de energía a través de la realización de trabajo mecánico, como la fuerza que se necesita para mover un objeto. Esta transferencia de energía se produce cuando se aplica una fuerza a un objeto y se mueve una distancia.
(e) Transmisión de energía eléctrica: es la transferencia de energía eléctrica a través de un conductor eléctrico, como un cable. Esta transferencia de energía se produce cuando se mueven electrones a través del conductor.
(f) Transferencia de energía nuclear: es la transferencia de energía a través de reacciones nucleares, como la fisión nuclear y la fusión nuclear. Esta transferencia de energía se produce cuando se libera la energía almacenada en los núcleos de los átomos.
Estos mecanismos de transferencia de energía tienen aplicaciones prácticas en muchos aspectos de nuestra vida cotidiana, como la calefacción y refrigeración de edificios, la generación de energía eléctrica, la iluminación y el transporte. Es importante entender cómo se transfieren y se transforman la energía para poder utilizarla de manera eficiente y sostenible.
(3.2) Energía en si misma
La energía se define como la capacidad de un sistema para realizar trabajo o producir cambio. Es una propiedad de los objetos o sistemas que les permite realizar actividades y transformaciones en su entorno. A diferencia de los mecanismos de transferencia de energía, la energía en sí misma no se transfiere, sino que se almacena en diferentes formas y se convierte de una forma a otra. Tipos de energía en sí misma:
(a) Energía térmica: es la energía asociada con la temperatura de un objeto. Se transfiere de un objeto a otro a través de la conducción, convección y radiación. Se utiliza en sistemas de calefacción, refrigeración y producción de energía eléctrica.
(b) Energía cinética: es la energía asociada con el movimiento de un objeto. Depende de la masa y la velocidad del objeto. Se utiliza en la producción de energía eléctrica a partir de turbinas hidráulicas, eólicas y de vapor.
(c) Energía potencial: es la energía asociada con la posición o el estado de un objeto. Depende de la altura, la fuerza gravitatoria y la elasticidad. Se utiliza en la energía hidroeléctrica, energía nuclear y energía geotérmica.
(d) Energía eléctrica: es la energía asociada con la carga eléctrica en movimiento. Se utiliza en la producción y distribución de energía eléctrica, así como en la tecnología de baterías y células solares.
(e) Energía química: es la energía almacenada en enlaces químicos entre átomos y moléculas. Se utiliza en la producción de energía a partir de combustibles fósiles, baterías y células de combustible.
(f) Energía nuclear: es la energía almacenada en el núcleo de un átomo. Se libera en procesos de fisión nuclear y fusión nuclear. Se utiliza en la producción de energía nuclear y en aplicaciones militares.
Estos son algunos de los tipos de energía en sí mismos con sus características y aplicaciones, pero existen muchos otros tipos como la energía hidráulica, la energía solar, la energía de biomasa, la energía del viento, entre otros.
(3.3) Energía cinética y potencial
Los objetos, ya sean pelotas de béisbol o moléculas, pueden poseer energía cinética, la energía del movimiento. La magnitud de la energía cinética, Ek, de un objeto depende de su masa, m y velocidad, u, no usaremos la v en el contexto químico debido a que se usa para el coeficiente estequiométrico:

Por lo tanto, la energía cinética de un objeto aumenta a medida que aumenta su velocidad. Por ejemplo, un automóvil que se mueve a 55 mi/h tiene mayor energía cinética que a 25 mi/h. Para una velocidad dada, la energía cinética aumenta al aumentar la masa. Por lo tanto, un camión grande que viaja a 55 mi/h tiene mayor energía cinética que un sedán pequeño que viaja a la misma velocidad porque el camión tiene la mayor masa.
En química, estamos interesados en la energía cinética de los átomos y las moléculas. Aunque son demasiado pequeñas para ser vistas, estas partículas tienen masa y están en movimiento y, por lo tanto, poseen energía cinética.
Todos los demás tipos de energía, la energía almacenada en un resorte estirado, en un peso sostenido sobre su cabeza o en un enlace químico, por ejemplo, son energía potencial. Un objeto tiene energía potencial en virtud de su posición con respecto a otros objetos. La energía potencial es, en esencia, la energía "almacenada" que surge de las atracciones y repulsiones que un objeto experimenta en relación con otros objetos.
Estamos familiarizados con muchos casos en los que la energía potencial se convierte en energía cinética. Por ejemplo, piense en un ciclista en la cima de una colina Figura 3.1. Debido a la fuerza de gravedad atractiva, la energía potencial del ciclista y su bicicleta es mayor en la cima de la colina que en la parte inferior. Como resultado, la bicicleta se mueve fácilmente cuesta abajo con mayor velocidad. Al hacerlo, la energía potencial se convierte en energía cinética. La energía potencial disminuye a medida que la bicicleta rueda cuesta abajo, pero su energía cinética aumenta a medida que aumenta la velocidad (Eq 3.1). Este ejemplo ilustra que las formas de energía son interconvertibles.

Figura 3.1. Energía potencial y energía cinética. La energía potencial inicialmente almacenada en la bicicleta inmóvil y el ciclista en la cima de la colina se convierte en energía cinética a medida que la bicicleta se mueve cuesta abajo y pierde energía potencial.
Las fuerzas gravitacionales juegan un papel insignificante en la forma en que los átomos y las moléculas interactúan entre sí. Las fuerzas que surgen de las cargas eléctricas son más importantes cuando se trata de átomos y moléculas. Una de las formas más importantes de energía potencial en química es la energía potencial electrostática, Ep,e, que surge de las interacciones entre partículas cargadas. Esta energía es proporcional a las cargas eléctricas en los dos objetos que interactúan, Qi y Qj, e inversamente proporcional a la distancia, ∆x, que los separa.

En esta ecuación, k es la constante de proporcionalidad, 8.99×109 J m / C2, que relaciona las unidades de energía con las unidades de carga eléctrica y su distancia de separación. (C) es el culombio, una unidad de carga eléctrica, y (J) es el julio, una unidad de energía que discutiremos pronto. A nivel molecular, las cargas eléctricas Qi y Qj son típicamente del orden de magnitud de constante de carga elemental: qe = 1.60217662 × 10-19 C.
La ecuación (Eq 3.2) muestra que la energía potencial electrostática llega a cero cuando ∆x se vuelve infinita. Por lo tanto, el cero de energía potencial electrostática se define como la separación infinita de las partículas cargadas.

Figura 3.2. Energía potencial electrostática. A distancias de separación finitas para dos partículas cargadas, Eel es positiva para cargas similares y negativa para cargas opuestas. A medida que las partículas se alejan más, su energía potencial electrostática se acerca a cero.
La Figura 3.2 ilustra cómo se comporta Eel a medida que cambia la distancia entre dos cargas. Cuando Qi y Qj tienen el mismo signo (por ejemplo, ambos positivos), las dos partículas cargadas se repelen entre sí, y una fuerza repulsiva las separa. En este caso, la anguila es positiva, y la energía potencial disminuye a medida que las partículas se separan más. Cuando Qi y Qj tienen signos opuestos, las partículas se atraen entre sí, y una fuerza atractiva las atrae entre sí. En este caso, Eel sea negativa y la energía potencial aumenta (se vuelve menos negativa) a medida que las partículas se separan.
Uno de nuestros objetivos en química es relacionar los cambios de energía observados en el mundo macroscópico con la energía cinética o potencial de las sustancias a nivel molecular. Muchas sustancias, como los combustibles, por ejemplo, liberan energía cuando reaccionan. La energía química de un combustible se debe a la energía potencial almacenada en las disposiciones de sus átomos.
Cuando se quema un combustible, esta energía química se convierte en energía térmica, energía asociada con la temperatura. El aumento de la energía térmica surge del aumento del movimiento molecular y, por lo tanto, del aumento de la energía cinética.
Unidades de energía
La unidad SI para la energía es el joule (pronunciado "yul"), J, en honor de James Joule (1818-1889), un científico británico que investigó el trabajo y el calor: 1 J = 1 kg m2/s2. Debido a que un joule no es una gran cantidad de energía, a menudo usamos kilojoules (kJ) para discutir las energías asociadas con las reacciones químicas.
Tradicionalmente, los cambios de energía que acompañan a las reacciones químicas se han expresado en calorías, una unidad no perteneciente al SI que todavía se usa ampliamente en química, biología y bioquímica. Una caloría (cal) se definió originalmente como la cantidad de energía requerida para elevar la temperatura de 1 g de agua de 14.5 a 15.5 ° C.
(a) Igualdad caloría menor o caloría mecánica a julios: 1 cal = 4.184 J.
(b) Igualdad unidades de gases a julios: 1 atm L=101.3 J.
(c) Igualdad caloría mayor o caloría nutricional a julios: 1 Cal = 103 cal = 1 kcal.
Una unidad de energía relacionada utilizada en nutrición es la Caloría nutricional (tenga en cuenta la C mayúscula).
4. Sistema, alrededores y calor
4. Sistema, alrededores y calor
Al analizar los cambios de energía, debemos centrarnos en una parte limitada y bien definida del universo para realizar un seguimiento de los cambios de energía que ocurren. La porción que seleccionamos para estudio se llama sistema; todo lo demás se llama entorno. Cuando estudiamos el cambio de energía que acompaña a una reacción química en un laboratorio, los reactivos y productos constituyen el sistema. El contenedor y todo lo que está más allá se consideran los alrededores.

Figura 4.1. Tipos de sistema por flujo de materia y energía.
Los sistemas pueden estar abiertos, cerrados o aislados. Un sistema abierto es aquel en el que la materia y la energía pueden intercambiarse con el entorno. Una olla descubierta de agua hirviendo en una estufa, es un sistema abierto: el calor entra al sistema desde la estufa y el agua se libera a los alrededores como vapor.
Los sistemas que podemos estudiar más fácilmente en termoquímica se denominan sistemas cerrados, sistemas que pueden intercambiar energía, pero no comparten materia con su entorno. Por ejemplo, considere una mezcla de gas hidrógeno, y gas oxígeno en un cilindro equipado con un pistón.

Figura 4.2. Sistema cerrado. Un sistema cerrado es un sistema termodinámico en el cual no hay transferencia de materia hacia adentro ni hacia afuera del sistema, pero puede haber intercambio de energía con el entorno en forma de calor o trabajo. En este caso el trabajo será realizado desde o hacia el sistema por medio del émbolo móvil.
El sistema es solo el hidrógeno y el oxígeno; el cilindro, el pistón y todo lo que está más allá de ellos (incluidos nosotros) son los alrededores. Si los gases reaccionan para formar agua, se libera energía: 2H2(g) + O2(g) → 2H2 O(g) + energía.
Aunque la forma química de los átomos de hidrógeno y oxígeno en el sistema cambia por esta reacción, el sistema no ha perdido ni ganado masa, lo que significa que no ha intercambiado ninguna materia con su entorno. Sin embargo, puede intercambiar energía con su entorno en forma de trabajo y calor al elevar el pistón o calentar su frontera.
Un sistema aislado es aquel en el que ni la energía ni la materia pueden intercambiarse con el entorno. Un termo aislado que contiene café caliente se aproxima a un sistema aislado. Sin embargo, sabemos que el café finalmente se enfría, por lo que no está perfectamente aislado.
La Figura 4.3 ilustra las dos formas en que experimentamos cambios de energía en nuestra vida cotidiana: en forma de trabajo y en forma de calor. En la Figura 4.3a, el trabajo se realiza a medida que la energía se transfiere del brazo del lanzador a la pelota, dirigiéndola hacia la placa a alta velocidad. En la Figura 4.3b la energía se transfiere en forma de calor. Causar el movimiento de un objeto contra una fuerza y provocar un cambio de temperatura son las dos formas generales en que la energía puede transferirse dentro o fuera de un sistema.

Figura 4.3. Trabajo y calor, dos formas de energía. (a) El trabajo es la energía utilizada para provocar un movimiento de objeto. (b) El calor es la energía utilizada para aumentar la temperatura de un objeto.
Definimos trabajo, W (mayúscula), como la energía transferida cuando una fuerza mueve un objeto. Una fuerza es cualquier empuje o atracción ejercida sobre un objeto. La magnitud del trabajo es igual al producto de la fuerza, F, y la distancia, Δr, el objeto se mueve:

Realizamos trabajo, por ejemplo, cuando levantamos un objeto contra la fuerza de la gravedad. Si definimos el objeto como el sistema, entonces nosotros, como parte del entorno, estamos realizando un trabajo en ese sistema, transfiriéndole energía.
La otra forma en que se transfiere la energía es como calor. El calor es la energía transferida de un objeto más caliente a uno más frío. Una reacción de combustión, como la quema de gas natural ilustrada en la Figura 4.3b, libera la energía química almacenada en las moléculas del combustible. Si definimos las sustancias involucradas en la reacción como el sistema y todo lo demás como el entorno, encontramos que la energía liberada hace que la temperatura del sistema aumente. La energía en forma de calor se transfiere desde el sistema más caliente al más frío y nunca en sentido opuesto.
5. La primera ley de la termodinámica
5. La primera ley de la termodinámica
Hemos visto que la energía potencial de un sistema se puede convertir en energía cinética, y viceversa. También hemos visto que la energía puede transferirse de un lado a otro entre un sistema y sus alrededores en forma de trabajo y calor. Todas estas conversiones y transferencias proceden de acuerdo con una de las observaciones más importantes de la ciencia: la energía no se puede crear ni destruir. Cualquier energía que se pierde por un sistema debe ser ganada por los alrededores, y viceversa. Esta observación importante, que la energía se conserva, se conoce como la primera ley de la termodinámica. Para aplicar esta ley cuantitativamente, primero definamos la energía de un sistema con mayor precisión:
(5.1) Energía interna
La energía interna, E, de un sistema es la suma de todas las energías cinéticas y potenciales de los componentes del sistema:

Para el sistema de la Figura 4.2, por ejemplo, la energía interna incluye no solo los movimientos e interacciones de las moléculas de H2 y O2, sino también los movimientos e interacciones de sus núcleos y electrones componentes. Generalmente no conocemos el valor numérico de la energía interna de un sistema. En termodinámica, estamos principalmente interesados en el cambio de energía entre dos momentos o estados arbitrarios ΔE (y, como veremos, también en otras cantidades) que acompaña a un cambio en el sistema.
Imagine que comenzamos con un sistema con una energía interna inicial Eo,i. Luego, el sistema sufre un cambio, que podría implicar el trabajo realizado o la transferencia de calor. Después del cambio, la energía interna final del sistema es Ei. Definimos el cambio en la energía interna, denotado ΔEi (leer "delta E de i"), como la diferencia entre los estados inicial y final.

Generalmente no podemos determinar los valores reales de Eo,i y Ei para ningún sistema de interés práctico. Sin embargo, podemos determinar el valor de ΔEi experimentalmente aplicando la primera ley de la termodinámica.
Las cantidades termodinámicas como ΔEi tienen tres partes: (1) un número y (2) una unidad, que juntas dan la magnitud del cambio, y (3) un signo que da la dirección.
(a) Se produce un valor positivo de ΔEi cuando ΔEi > ΔEo,i, lo que indica que el sistema ha obtenido energía de su entorno.
(b) Se produce un valor negativo de ΔEi cuando ΔEi < ΔEo,i, lo que indica que el sistema ha perdido energía en su entorno.
Observe que estamos discutiendo los puntos de vista del sistema en lugar de los del entorno al discutir los cambios de energía. Sin embargo, debemos recordar que cualquier aumento en la energía del sistema va acompañado de una disminución en la energía del entorno, y viceversa.
En una reacción química que no involucra situaciones de equilibrio, el estado inicial del sistema se refiere a los reactivos y el estado final se refiere a los productos. En la reacción: 2H2(g) + O2(g) → 2H2O(l).
Por ejemplo, el estado inicial es 2H2(g) + O2(g) y el estado final es el 2H2O(l). Cuando el hidrógeno y el oxígeno forman agua a una temperatura dada, el sistema pierde energía en los alrededores. Debido a que el sistema pierde energía, la energía interna de los productos (estado final) es menor que la de los reactivos (estado inicial), y ΔE para el proceso es negativo.
(5.2) Cambio de energía, trabajo y calor
Un sistema puede intercambiar energía con su entorno de dos maneras generales: como calor o como trabajo. La energía interna de un sistema cambia en magnitud a medida que el calor se agrega o se elimina del sistema o cuando se realiza trabajo en el sistema. Si pensamos en la energía interna como la cuenta bancaria de energía del sistema, vemos que los depósitos o retiros se pueden hacer en forma de calor o en forma de trabajo. Los depósitos aumentan la energía del sistema (positivo ΔE), mientras que los retiros disminuyen la energía del sistema (negativo ΔE).
Podemos usar estas ideas para escribir una expresión algebraica útil de la primera ley de la termodinámica. Cuando un sistema sufre cualquier cambio químico o físico, el cambio que lo acompaña en la energía interna, ΔE, es la suma del calor agregado o liberado del sistema, Q, y el trabajo realizado en o por el sistema, W.

Cuando se agrega calor a un sistema o se realiza trabajo en un sistema, su energía interna aumenta. Por lo tanto,
(a) cuando el calor se transfiere al sistema desde los alrededores, Q tiene un valor positivo.
(b) cuando el trabajo se transfiere al sistema desde los alrededores, W tiene un valor positivo.
Agregar calor al sistema es como hacer un depósito en la cuenta de energía: la energía del sistema aumenta. Del mismo modo, cuando el entorno realiza el trabajo en el sistema, W tiene un valor positivo. Por el contrario, tanto el calor perdido por el sistema hacia los alrededores como el trabajo realizado por el sistema en los alrededores tienen valores negativos; es decir, reducen la energía interna del sistema. Son retiros de energía y reducen la cantidad de energía en la cuenta del sistema.
(5.3) Procesos endotérmicos y exotérmicos
Debido a que la transferencia de calor hacia y desde el sistema es central para nuestra discusión en este capítulo, tenemos una terminología especial para indicar la dirección de la transferencia. Cuando ocurre un proceso en el cual el sistema absorbe calor, el proceso se llama endotérmico (endo significa "dentro"). Durante un proceso endotérmico, como la fusión del hielo, el calor fluye hacia el sistema desde su entorno. Si nosotros, como parte de los alrededores, tocamos un recipiente en el que se derrite el hielo, el recipiente se siente frío porque el calor ha pasado de nuestra mano al recipiente.
Un proceso en el que el sistema pierde calor se llama exotérmico (exo significa "fuera de"). Durante un proceso exotérmico, como la combustión de gasolina, el calor sale del sistema hacia los alrededores.
(5.4) Funciones de estado
Aunque generalmente no tenemos forma de conocer el valor preciso de la energía interna de un sistema, , sí tiene un valor fijo para un conjunto dado de condiciones. Las condiciones que influyen en la energía interna incluyen la temperatura y la presión. Además, la energía interna de un sistema es proporcional a la cantidad total de materia en el sistema porque la energía es una propiedad extensiva.
Supongamos que definimos nuestro sistema como 50 g de agua a 25 °C. El sistema podría haber alcanzado este estado enfriando 50 g de agua de 100°C a 25 °C o derritiendo 50 g de hielo y posteriormente calentando el agua a 25 °C. La energía interna del agua a 25 °C es la misma, en cualquier caso. La energía interna es un ejemplo de una función de estado, una propiedad de un sistema que se determina especificando la condición o estado del sistema (en términos de temperatura, presión, etc.). El valor de una función de estado depende solo del estado actual del sistema, no de la ruta que el sistema tomó para alcanzar ese estado. Como E es una función de estado, ∆E depende solo de los estados inicial y final del sistema, no de cómo se produce el cambio.
Una analogía puede ayudarlo a comprender la diferencia entre las cantidades que son funciones de estado y las que no lo son. Suponga que conduce desde una ciudad A, que está a 1551 metros sobre el nivel del mar, hasta una ciudad B, que está a 2640 metros sobre el nivel del mar. No importa qué ruta tome, el cambio de altitud es 1089 metros. La distancia que recorre, sin embargo, depende de su ruta. La altitud es análoga a una función de estado porque el cambio de altitud es independiente de la ruta tomada. La distancia recorrida no es una función de estado.
Algunas cantidades termodinámicas, como E, son funciones de estado. Otras cantidades, como Q y W, no lo son. Esto significa que, aunque ∆E = Q + W no depende de cómo se produce el cambio, las cantidades específicas de calor y trabajo dependen de la forma en que se produce el cambio. Por lo tanto, si cambia la ruta por la cual un sistema pasa de un estado inicial a un estado final aumenta el valor de Q, ese cambio de ruta también disminuirá el valor de W exactamente en la misma cantidad. El resultado es que ∆E es igual para las dos rutas.
Podemos ilustrar este principio utilizando una batería de linterna como nuestro sistema. A medida que la batería se descarga, su energía interna disminuye a medida que la energía almacenada en la batería se libera a los alrededores.

Figura 5.1. La energía interna es una función de estado, pero el calor y el trabajo no lo son. (a) Una batería unida a un cable de calefacción pierde energía en los alrededores solo como calor; No se realiza ningún trabajo. (b) Una batería descargada a través de un motor pierde energía como trabajo (para hacer que el ventilador gire) y también pierde algo de energía como calor. El valor de ∆E es el mismo para ambos procesos, aunque los valores de Q y W en (a) son diferentes de los de (b).
En la Figura 5.1, consideramos dos formas posibles de descargar la batería a temperatura constante. Si un cable de calefacción se une a la batería, no se realiza ningún trabajo porque nada se mueve contra una fuerza. Toda la energía perdida de la batería está en forma de calor. (El cable se calienta y libera calor a los alrededores.) Si la batería se usa para hacer girar el motor, la descarga produce trabajo. Se libera algo de calor, pero no tanto como cuando la batería está en un calefactor. Vemos que las magnitudes de Q y W deben ser diferentes para estos dos casos. Sin embargo, si los estados inicial y final de la batería son idénticos en los dos casos, entonces ∆E = Q + W debe ser el mismo en ambos casos porque E es una función de estado. Recuerde: ∆E depende solo de los estados inicial y final del sistema, no de la ruta específica tomada del estado inicial al final.
6. Entalpía
6. Entalpía
Los cambios químicos y físicos que ocurren a nuestro alrededor, como la fotosíntesis en las hojas de una planta, la evaporación del agua de un lago o una reacción en un vaso abierto en un laboratorio, ocurren bajo la presión esencialmente constante de la atmósfera de la Tierra. Estos los cambios pueden provocar la liberación o absorción de calor y pueden ir acompañados de un trabajo realizado por el sistema o sobre él. Al explorar estos cambios, es útil tener una función termodinámica que es una función de estado y se relaciona principalmente con el flujo de calor. En condiciones de presión constante, una cantidad termodinámica llamada entalpía (del griego enthalpeína, "calentar") cumple dicha función.
La entalpía, que denotamos con el símbolo H, se define como la energía interna más el producto de la presión, P y el volumen, V, del sistema:

Esto se debe a que el ambiente mismo posee una energía subyacente que resulta de multiplicar la presión por el volumen. Recuerde que esto está vinculado a la relación entre julios atmósferas y litros: 101.3 J = 1 atm L.
Al igual que la energía interna Ei, tanto Pi como Vi son funciones de estado: dependen solo del estado actual del sistema y no de la ruta tomada a ese estado. Debido a que la energía, la presión y el volumen son funciones de estado, la entalpía también es una función de estado.
(6.1) Presión volumen y trabajo
Para comprender mejor la importancia de la entalpía, recuerde de la Ecuación : ∆E = Q + W; que ∆E involucra no solo el calor Q agregado o eliminado del sistema, sino también el trabajo realizado por o en el sistema. Más comúnmente, el único tipo de trabajo producido por cambios químicos o físicos abiertos a la atmósfera es el trabajo mecánico asociado con un cambio en el volumen. Por ejemplo, cuando la reacción de zinc metálico con solución de ácido clorhídrico: Zn(s) + 2H+(aq) → Zn2+ (aq) + H2(g); se ejecuta a presión constante en el aparato ilustrado en la Figura 6.1, el pistón se mueve hacia arriba o hacia abajo para mantener una presión constante en el recipiente.

Figura 6.1. Un sistema que funciona en su entorno, este es mi experimentales una modificación de el manómetro de Huygens, en la cual la presión del sistema va a cambiar debido a la reacción química entre el cine metálico y el ácido clorhídrico, el cual al liberar hidrógeno, aumenta la presión, y al aumentar la presión se genera trabajo sobre los alrededores.
Si suponemos por simplicidad que el pistón no tiene masa, la presión en el aparato es la misma que la presión atmosférica. A medida que avanza la reacción, se forma gas H2 y el pistón sube. El gas dentro del matraz está haciendo trabajo en los alrededores al levantar el pistón contra la fuerza de la presión atmosférica.
El trabajo involucrado en la expansión o compresión de gases se llama trabajo presión-volumen (trabajo P – V). Cuando la presión es constante en un proceso, como en nuestro ejemplo anterior, el signo y la magnitud del trabajo de presión-volumen están dados por: Wi = -Pi ∆Vi; donde es presión y: ∆Vi = Vi - Vo,i; es el cambio en el volumen del sistema. La presión Pi es siempre un número positivo o cero. Si el volumen del sistema se expande, entonces Vi también es positivo. El signo negativo en Wi = -Pi ∆Vi es necesario para cumplir con la convención de signos para W.
(a) Cuando un gas se expande, el sistema funciona en los alrededores, como lo indica un valor negativo de W. Por otro lado…
(b) Cuando el gas está comprimido, ∆V es negativo (el volumen disminuye), y Wi = -Pi ∆Vi indica que W es positivo, lo que significa que el trabajo se realiza en el sistema por los alrededores.
Las unidades de trabajo obtenidas mediante el uso Wi = -Pi ∆Vi serán las de presión (generalmente atm) multiplicadas por las de volumen (generalmente L). Para expresar el trabajo en la unidad de julios más familiar, utilizamos el factor de conversión 1 L atm = 101.3 J.
(6.2) El cambio de energía interna

|
Tenga en cuenta que en este enunciado la presión y los volúmenes corresponden al sistema, por lo que bien podríamos tener un sistema de mezcla de varios gases, y por ende, debemos tener en cuenta la posibilidad de aplicar la ley de presiones parciales de Dalton.
(6.3) Trabajo hecho por una reacción química
Si tenemos una reacción química tendremos que el volumen inicial estará dado por la suma de volúmenes de los reactivos, mientras que el volumen final por la suma de volúmenes de los productos. Así que ¿Cuál es la función que permite determinar el trabajo de una reacción química donde hay gases?

|
7. Entalpía de la reacción
7. Entalpía de la reacción
El cambio de entalpía que acompaña a una reacción se llama entalpía de reacción o calor de reacción.

donde ∆H→ indica que es una medida para la reacción en sentido directo, aunque lo común es expresarlo sin ningún subíndice ∆H, lo cual indica que es una entalpía que no está asociada a una sustancia concreta. Cuando damos un valor numérico para ∆H, debemos especificar la reacción involucrada. Por ejemplo, cuando 2 mol de H2(g) se queman para formar 2 mol de H2O(g) a una presión constante, el sistema libera 483.6 kJ de calor. Podemos resumir esta información como:

(a) si el signo negativo para ∆H nos dice que esta reacción es exotérmica.
(b) si el signo positivo para ∆H nos dice que esta reacción es endotérmica.
Observe que ∆H se informa al final de la ecuación balanceada, sin especificar explícitamente las cantidades de productos químicos involucrados.
(7.1) Avance de la reacción
Al igual que como sucede con la masa m y la masa molar M para las entalpias, tendremos una dicotomía entre la entalpía simple ΔH y la entalpía estándar ΔHo, que, aunque relacionadas, tendrán unidades diferentes. La entalpía de la reacción ΔH es una propiedad extensiva, por lo tanto, a mayor cantidad de sustancia n de reactivos y productos, ΔH va a ser mayor la energía requerida o generada por la reacción. Dado que la entalpía de la reacción es un parámetro que depende de la reacción como un todo o de las cantidades de sustancia, puede vincularse al avance de la reacción (Baeza-Baeza & García-Alvarez-Coque, 2014; Canagaratna, 2000; Croce, 2002; De Donder & Van Rysselberghe, 1936; García-García, 2021; García García, 2020; Garst, 1974; Hanyak Jr, 2014; IUPAC, McNaught, & Wilkinson, 2019; Moretti, 2015; Mousavi, 2018; Wikipedia, 2019).
Una reacción química puede repetirse una cantidad arbitraria de veces, sin embargo, la entalpía estándar ΔHo siempre está fijada a una cantidad de reacción específica que denominaremos como el avance de la reacción estándar ξu. En consecuencia, la entalpía estándar ΔHo va a ser una propiedad intensiva para una reacción química particular fijada a ξu.
Recuerde que el avance de la reacción ξ determina cuántas veces de Avogadro se repite un evento de reacción y para el avance de reacción estándar, vamos a asumir que la reacción se repite una un número de Avogadro, es decir, lo lo que es lo mismo, se repite una mol de veces, por ende ξu = 1 mol. Dado lo anterior,
(a) las entalpias simples ΔH van a estar medidas en unidades de energía como J, mientras que…
(b) las entalpías estándar ΔHo van a estar medidas en unidades de energía ponderadas a la cantidad de veces que se repite la reacción es decir J/mol.
De lo anterior también se desprende que la entalpía de la reacción va a ser igual al producto entre el avance de la reacción y la entalpía estándar.
Demostración: Hallar la ecuación para convertir entre entalpía estándar J/mol y entalpía específica J/g // Pulse aquí

|
Eq 7.3 Entalpía de la reacción en términos de la entalpía estándar y el avance de la reacción
Eq 7.4 Entalpía de la reacción en términos de la entalpía estándar, la cantidad de sustancia y el número estequiométrico de una sustancia clave.
Eq 7.5 Entalpía de la reacción en términos de la entalpía estándar, la masa y masa molar de sustancia y el número estequiométrico de una sustancia clave.
Eq 7.6 Entalpía de la reacción en términos de la entalpía estándar específica para una sustancia en la reacción, y la cantidad en moles de esa sustancia.
Eq 7.7 Entalpía de la reacción en términos de la entalpía estándar específica para una sustancia en la reacción, la masa y masa molar de esa sustancia.
Eq 7.8 Entalpía estándar de la reacción en términos de la entalpía estándar específica para una sustancia en la reacción, y el número estequiométrico de la sustancia.
Eq 7.9 Entalpía de la reacción específica a una sustancia en términos de la entalpía estándar, la masa molar de sustancia y el número estequiométrico de una sustancia clave.
Eq 7.10 Avance de la reacción para reacciones químicas no reversibles o cuya reversibilidad es despreciable.
Eq 7.11. Entalpía estándar de la sustancia en términos de la entalpía específica de la sustancia y la masa molar
(7.2) Propiedades de la entalpía de la reacción
Las siguientes pautas son útiles cuando se usan ecuaciones termoquímicas y diagramas de entalpía:
La entalpía es una propiedad extensiva
La magnitud de ∆H es proporcional a la cantidad de reactivo consumido en el proceso o a la cantidad de producto generado, aunque visto desde el punto de vista de la reacción, es directamente proporcional al avance de la reacción, es decir, cuantas veces se repite un solo evento de reacción (Eq 7.4).
Las ecuaciones reversas tienen entalpías opuestas
Cuando revertimos una reacción, revertimos los roles de los productos y los reactivos. En estos casos es donde es conveniente usar la notación ∆H→ para la reacción directa y ∆H← para la reacción reversa, en lugar de solo ∆H.

La entalpía depende de los estados de la materia
La entalpía es una variable que no solo depende de la cantidad de sustancia y la identidad de la sustancia, también depende del estado de la materia y la presión, por lo que dos reacciones con ecuaciones iguales, pero con estados de materia diferentes y alguna de las sustancias producto o reactivo tendrá un valor de entalpía para toda la reacción diferenciado.
En muchas situaciones encontraremos valioso conocer el signo y la magnitud del cambio de entalpía asociado con un proceso químico dado. Como vemos en las siguientes secciones, ∆H puede determinarse directamente por experimento o calcularse a partir de cambios de entalpía conocidos de otras reacciones.
(7.3) Relación entre cambio de entalpía y cambio de energía interna
Frecuentemente los productos de una reacción no se limitan únicamente a la generación de calor, también involucran la generación de gases a muy altas presiones que generan un empuje expansivo importante, y por ende ejercen trabajo a los alrededores. De allí que la ley de la conservación de la energía implique la suma de la energía de la reacción más la energía por trabajo mecánico.

|
8. Capacidad de calorífica y calor específico
8. Capacidad de calorífica y calor específico
El valor de ∆H se puede determinar experimentalmente midiendo el flujo de calor que acompaña a una reacción a presión constante. Por lo general, podemos determinar la magnitud del flujo de calor midiendo la magnitud del cambio de temperatura que produce el flujo de calor. La medida del flujo de calor es calorimetría; Un dispositivo utilizado para medir el flujo de calor es un calorímetro.
Cuanto más calor gana un objeto, más caliente se pone. Todas las sustancias cambian de temperatura cuando se calientan, pero la magnitud del cambio de temperatura producido por una cantidad dada de calor varía de una sustancia a otra. El cambio de temperatura experimentado por un objeto cuando absorbe una cierta cantidad de calor está determinado por su capacidad de calor, denotada C. La capacidad de calor de un objeto es la cantidad de calor requerida para elevar su temperatura en 1 K (o 1 °C). Cuanto mayor es la capacidad de calor, mayor es el calor requerido para producir un aumento dado de temperatura.
Para las sustancias puras, la capacidad calorífica generalmente se da para una cantidad específica de la sustancia.
(a) La capacidad calorífica (Ci) (Unidades J/°C o J/K) es el límite cuando el cambio de temperatura tiende a cero de la diferencia de calor sobre la diferencia de temperatura, aunque en la práctica es el calor asociado a la variación de un grado de unidad de temperatura.
(b) El calor específico de un mol de una sustancia se llama calor específico molar, (Cmi) (Unidades J/°C-mol o J/K-mol).
(c) El calor específico de un gramo de una sustancia se denomina calor específico, (Csi) (Unidades J/°C-g o J/K-g).

|
Los calores específicos de muchas sustancias notables se encuentran indexadas en tablas estandarizadas, como la consignada en este enlace para metales y metaloides.
(8.1) El cambio de temperatura
Si bien una temperatura en kelvin es en términos absolutos diferente al valor que tenemos a la temperatura en grados Celsius, y por ende tenemos que sumar o restar los 273.15 y ajustar por medio de un factor de conversión las unidades correctas, la cuestión es diferente cuando tenemos un cambio de temperatura ∆T. Debido a que la magnitud de los grados Celsius y las unidades kelvin son exactamente iguales, la diferencia entre dos temperaturas entre las dos escalas va a tener el mismo valor, por ende la diferencia de temperaturas en grados Celsius va a ser igual a la diferencia de temperaturas en kelvin: ∆T°C = ∆TK. Esta conversión de plumazo no es válida con la escala Fahrenheit, debido precisamente a que la magnitud de 1 °F es diferente a la magnitud de 1 k o 1° Celsius.
(8.2) Calor absorbido o emitido por la masa de un cuerpo
Tenga en cuenta que:
Demostración: Encuentre una función que permita calcular el calor en términos del calor específico, la masa y el cambio de temperatura // Pulse aquí.

El calor específico, Csi, de una sustancia se puede determinar experimentalmente midiendo el cambio de temperatura, ∆T, que sufre una masa conocida m de la sustancia cuando gana o pierde una cantidad específica de calor Q:
(a) si el signo negativo para Q nos dice que esta reacción es exotérmica.
(b) si el signo positivo para Q nos dice que esta reacción es endotérmica.
Un cambio de temperatura en Kelvin es igual en magnitud al cambio de temperatura en grados Celsius. Por lo tanto, este calor específico para el agua también se puede informar como 4.18 J/g-°C = 4.18 J/(g °C), donde la unidad se pronuncia "Julios por gramo-grado Celsius". Debido a que los valores de calor específicos para una sustancia dada pueden variar ligeramente con la temperatura, la temperatura a menudo se especifica con precisión. El valor de 4.18 J / g-K que usamos aquí para el agua, por ejemplo, es para agua inicialmente a 14.5 °C. El calor específico del agua a esta temperatura se usa para definir las calorías al valor dado cal = 4.184 J exactamente.
Por lo tanto, podemos calcular la cantidad de calor que una sustancia gana o pierde usando su calor específico junto con su masa medida y el cambio de temperatura. Los calores específicos dependen directamente de la identidad y cantidad de la sustancia, por lo que son en términos prácticos, constantes que deben consultarse en tablas, a menos que el enunciado del ejercicio los de o los pida.
Tabla 8.1. Calores específicos de algunas sustancias a 298 K.

(8.3) Calor compartido por dos cuerpos y temperatura de equilibrio
Ahora que ya sabemos calcular el calor absorbido o emitido por un solo cuerpo, abordaremos el problema de dos cuerpos que permiten el flujo de calor desde el cuerpo más caliente al cuerpo más frio hasta alcanzar el punto de equilibrio que corresponde a la temperatura de equilibrio. Por ende, ¿Cuál es la función que permite hallar dicha temperatura de equilibrio? La cosa es que esta pregunta implica un diseño experimental denominado calorímetro, y por ende, responderemos esta pregunta y sus consecuencias en las siguientes secciones.
9. Calorimetría a volumen constante
9. Calorimetría a volumen constante
Un tipo importante de reacción estudiada usando calorimetría es la combustión, en la cual un compuesto reacciona completamente con exceso de oxígeno. Las reacciones de combustión se estudian con mayor precisión utilizando un calorímetro de bomba:

Figura 9.1. Calorímetro de bomba. Ese calorímetro posee dos contenedores, el primero es el del calorímetro propiamente dicho que debe impedir el flujo de calor, y es el más externo. el segundo contenedor es la bomba, dónde se encuentra el reactivo combustible que va a reaccionar con el oxígeno u otra sustancia de ignición.
La sustancia a estudiar se coloca en una pequeña copa dentro de un recipiente sellado y aislado llamado bomba. La bomba, que está diseñada para soportar altas presiones, tiene una válvula de entrada para agregar oxígeno y cables eléctricos para iniciar la reacción. Después de colocar la muestra en la bomba, la bomba se sella y se presuriza con oxígeno. Luego se coloca en el calorímetro y se cubre con una cantidad de agua medida con precisión. La reacción de combustión se inicia haciendo pasar una corriente eléctrica a través de un cable fino en contacto con la muestra. Cuando el cable se calienta lo suficiente, la muestra se enciende.
El calor liberado cuando se produce la combustión es absorbido por el agua y los diversos componentes del calorímetro (que en conjunto forman el entorno), lo que hace que la temperatura del agua aumente. El cambio en la temperatura del agua causado por la reacción se mide con mucha precisión.
Dado que el proceso tiene lugar a volumen constante, el recipiente interno de reacción debe estar construido para resistir la alta presión resultante del proceso de combustión, que equivale a una explosión confinada. El recipiente se suele llamar "bomba" y la técnica se conoce como calorimetría de bomba. La reacción se inicia descargando un condensador a través de un alambre delgado que enciende la mezcla.
Debido a que el recipiente mismo hace parte del sistema que alcanza un equilibrio término, su calor específico deberá calcularse previamente en un procedimiento de estandarización
(9.1) Estandarizando el calor específico del calorímetro con el calor de reacción
Para calcular el calor de combustión a partir del aumento de temperatura medido, debemos conocer la capacidad calorífica total del calorímetro más el agua involucrada. Esta cantidad se determina quemando una muestra que libera una cantidad conocida de calor y midiendo el cambio de temperatura con todo y agua. La capacidad calorífica calculada de este modo la denominaremos capacidad calorífica estándar del calorímetro Cº. (incluyendo el agua). La notación estándar o inicial del calor de reacción y de la temperatura implican que son valores conocidos, mediante una reacción química ya conocida. Una de estas reacciones de estandarización es la del ácido benzoico.
(9.2) Ecuaciones
Una vez que tenemos el calor específico estándar, podemos deducir las fórmulas para el calorímetro de bomba, pero teniendo en cuenta que tendremos dos seres de funciones, las simplificadas y las exactas. Debido a que las reacciones en un calorímetro de bomba se llevan a cabo a volumen constante, el calor transferido corresponde al cambio en la energía interna, ∆E, en lugar del cambio en la entalpía, ∆H. Sin embargo, para la mayoría de las reacciones, la diferencia entre ∆E y ∆H es muy pequeña, aunque igual determinaremos funciones para ambos casos. Adicionalmente obtendremos cuatro tipos de entalpías diferentes.
(a) Entalpía de la reacción en unidades de J o kJ
(b) Entalpía estándar de la reacción en unidades de J/mol o kJ/mol, pero en este caso el mol corresponde al avance de la reacción completa y no a una sustancia particular, debido a las confusiones de este concepto, es que se convierte a:
(c) Entalpía de la reacción ajustada a la cantidad de sustancia clave (calor de combustión de una sustancia en moles) J/mol o kJ/mol, en este caso el mol si representa a una sustancia concreta.
(d) Entalpía de la reacción ajustada a la masa de sustancia clave (calor de combustión de una sustancia en gramos) J/g o kJ/g, donde g representa a una sustancia concreta.

Eq 9.1. Calor de la reacción con respecto a un calorímetro.
Eq 9.2. Calor de dos reacciones (ri) reacción problema o incógnita y (re) reacción de estandarización.
Eq 9.3. Calor estándar para una mol de reacción en términos del calor específico del calorímetro.
Eq 9.4. Calor estándar para una mol de reacción en términos de una reacción de estandarización
Eq 9.5. Entalpía específica molar de una sustancia en términos de la capacidad calorífica de un calorímetro
Eq 9.6. Entalpía específica molar de una sustancia en términos de una reacción de estandarización.
Eq 9.7. Entalpía específica a masa de una sustancia en términos de la capacidad calorífica de un calorímetro
Eq 9.8. Entalpía específica a masa de una sustancia en términos de una reacción de estandarización
Eq 9.9. Entalpía de la reacción con respecto al calorímetro y el efecto de cambio en cantidad de gas.
Eq 9.10. Entalpía estándar de la reacción con respecto al calorímetro y el efecto de cambio en cantidad de gas.
Eq 9.11. Entalpía estándar de la reacción con respecto a moles de una sustancia, al calorímetro y el efecto de cambio en cantidad de gas.
Eq 9.12. Entalpía estándar de la reacción con respecto a gramos de una sustancia, al calorímetro y el efecto de cambio en cantidad de gas.
Eq 9.13. Entalpía estándar de la reacción con respecto a moles de una sustancia, una reacción problema y una reacción de estandarización y el efecto de cambio en cantidad de gas.
10. Calorimetría a presión constante
10. Calorimetría a presión constante
Las técnicas y equipos empleados en calorimetría dependen de la naturaleza del proceso que se estudia. Para muchas reacciones, como las que ocurren en solución, es fácil controlar la presión para que se mida ∆H directamente.

Figura 10.1. Calorímetro de taza de café. Este simple aparato se usa para medir los cambios de temperatura de las reacciones a presión constante. Este es el tipo de calorímetro más común para los experimentos escolares.
Aunque los calorímetros utilizados para trabajos de alta precisión son instrumentos de precisión, un simple calorímetro de "taza de café"; (Figura 10.1) se usa a menudo en laboratorios de química general para ilustrar los principios de la calorimetría. Debido a que el calorímetro no está sellado, la reacción ocurre bajo la presión esencialmente constante de la atmósfera.
(10.1) Para un cuerpo inerte
La calorimetría para un cuerpo inerte se fundamenta en el flujo de calor entre dos cuerpos, uno incógnita o variable, y el otro el dato o conocido, que generalmente es agua. El punto es que el cuerpo inerte NO reacciona químicamente con el agua, y solo comparte físicamente calor con esta. Ahora pasemos al modelo matemático:
Podemos extraer las siguientes funciones clave.

Eq 10.1. Capacidad calorífica del cuerpo incógnita
Eq 10.2. Masa del cuerpo incógnita
Eq 10.3. Cambio de temperatura que sufre el cuerpo incógnita.
Eq 10.4. Término (x) o radio doble capacidad calorífica-masa incógnita-dato
Eq 10.5. Temperatura de equilibrio entre dos cuerpos
Eq 10.6. Temperatura inicial de la sustancia dato.
Eq 10.7. Temperatura inicial de la sustancia incógnita.
Como se mencionó en la demostración, la sustancia (d) generalmente es agua, pero no siempre es el caso, por lo que deberá adaptar las identidades para situaciones que implican diseños experimentales aparentemente diferentes, pero que encajan en el mismo modelo matemático. En otras ocasiones el enunciado se organiza de forma tal que el agua es la sustancia con menos términos conocidos, por lo que las definiciones se invierten.
(10.2) Para una reacción en agua
Al contrario de lo que podría esperarse, las ecuaciones para la calorimetría de reacciones a presión constante son más simples. En este caso el agua no es solo un cuerpo extra, es un solvente, lo cual nos lleva al hecho de que podría usarse otro solvente, por ende, definiremos esto para cualquier solvente (ii) y para el reactivo limitante (i).

|
Eq 10.8. Entalpía de la reacción en calorimetría.
Eq 10.9. Entalpía estándar de la reacción en calorimetría.
Eq 10.10. Entalpía estándar de una sustancia en calorimetría.
Eq 10.11. Diferencia de temperaturas del solvente en términos de la entalpía de la reacción.
Eq 10.12. Diferencia de temperaturas del solvente en términos de la entalpía estándar de la reacción.
Eq 10.13. Diferencia de temperaturas del solvente en términos de la entalpía estándar de una sustancia.
11. Ley de Hess
11. Ley de Hess
La entalpía o calor de reacción es básicamente un tipo de energía, y como toda energía está sujeta a la ley de la conservación de la energía, que es la suma ponderada de los aportes energéticos de los componentes de un sistema. Por ende, es posible calcular el ∆H para una reacción a partir de los valores tabulados de ∆H de otras reacciones más simples, o que en una suma lógica permite deducir la reacción problema. Por lo tanto, no es necesario realizar mediciones calorimétricas para todas las reacciones.
Debido a que la entalpía es una función de estado, el cambio de entalpía total, ∆H, asociado con cualquier proceso químico depende solo de la cantidad de materia que sufre cambios y de la naturaleza del estado inicial de los reactivos y el estado final de los productos. Esto significa que, si una reacción particular se lleva a cabo en un paso o en una serie de pasos r, la suma de los cambios de entalpía asociados con los pasos individuales ∑∆Hr debe ser la misma que el cambio de entalpía asociado con el proceso de un paso ∆H.
Todos los tipos de entalpía de reacción que hemos visto hasta ahora son susceptibles de ser modeladas mediante la ley Hess, en consecuencia, podemos tener enunciados para la entalpía de la reacción (Eq 11.1) y para la entalpía estándar de la reacción (Eq 11.2).

|
Eq 11.1. Ley de Hess para la entalpía de la reacción.
Eq 11.2. Ley de Hess para la entalpía estándar de la reacción.
Las entalpías de reacción son ponderables e invertibles, esto quiere decir que:
(a) si la cantidad de sustancia de un reactivo o producto en la reacción clave es n veces la presente en la reacción indirecta, toda la reacción indirecta incluida su entalpía se multiplica n veces.
(b) si un producto de la reacción problema se encuentra en los reactivos de las reacciones indirectas, invertimos la posición de reactivos y productos en la reacción indirecta y multiplicamos por -1 la entalpía de la reacción.
En algunas ocasiones, algún coeficiente de un reactivo o producto se calcula indirectamente a través de los coeficientes de las reacciones involucradas,

si el valor anterior es positivo significa que el número estequiométrico quedó del lado de los reactivos; por el contrario, si es un valor negativo significa que es un número o kilométrico que va del lado de los productos. Otro alternativo sería realizar la suma de las ecuaciones químicas completas, pero en este caso estaríamos sumando los coeficientes de todas las sustancias, incluso de aquellas de las cuales ya conocemos sus valores, porque los ajustamos manualmente al hacer el cálculo de la entalpía estándar o entalpía de la reacción.
12. Entalpías de formación
12. Entalpías de formación
Podemos utilizar los métodos que acabamos de comentar para calcular los cambios de entalpía para un gran número de reacciones a partir de valores de ∆Ho tabulados. Por ejemplo, existen tablas extensas de entalpías de vaporización (∆Ho para convertir líquidos en gases), entalpías de fusión (∆Ho para fundir sólidos), entalpías de combustión (∆Ho para quemar una sustancia en oxígeno), etc. Un proceso particularmente importante utilizado para tabular datos termoquímicos es la formación de un compuesto a partir de sus elementos constituyentes. El cambio de entalpía asociado con este proceso se denomina entalpía de formación (o calor de formación estándar), ∆Hfo, donde el subíndice f indica que la sustancia se ha formado a partir de sus elementos constituyentes.
La magnitud de cualquier cambio de entalpía depende de la temperatura, la presión y el estado (gas, líquido o forma cristalina sólida) de los reactivos y productos. Para comparar las entalpías de diferentes reacciones, debemos definir un conjunto de condiciones, llamado estado estándar, en el que se tabulan la mayoría de las entalpías. El estado estándar de una sustancia es su forma pura a presión atmosférica (1 atm) y la temperatura de interés, que generalmente elegimos ser 298 K o 25°C. El cambio de entalpía estándar de una reacción se define como el cambio de entalpía cuando todos los reactivos y productos están en sus estados estándar. Denotamos un cambio de entalpía estándar como ∆Ho, donde el superíndice 0 indica condiciones de estado estándar.
La entalpía estándar de formación de un compuesto, ∆Hf,io, es el cambio en la entalpía para la reacción que forma un mol del compuesto a partir de sus elementos con todas las sustancias en sus estados estándar. Por lo general, informamos valores de ∆Hf,io a 298 K. Si un elemento existe en más de una forma en condiciones estándar, la forma más estable del elemento se usa generalmente para la reacción de formación. Por ejemplo, la entalpía estándar de formación para el etanol, C2H5OH, es el cambio de entalpía para la reacción.
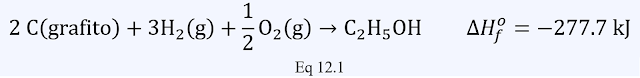
La fuente elemental de oxígeno es O2, no O u O3, porque O2 es la forma estable de oxígeno a 298 K y presión atmosférica. De manera similar, la fuente elemental de carbono es el grafito y no el diamante porque el grafito es la forma más estable (de menor energía) a 298 K y presión atmosférica. Asimismo, la forma más estable de hidrógeno en condiciones estándar es H2(g). En este enlace encontrará una tabla de entalpías estándar de formación para sustancias comunes.
Por definición, la entalpía estándar de formación de la forma más estable de cualquier elemento es cero porque no se necesita reacción de formación cuando el elemento ya se encuentra en su estado estándar. Por lo tanto, los valores de ∆Hf,io para C (grafito), H2(g), O2(g) y los estados estándar de otros elementos son cero por definición.
Las entalpías estándar de formación no necesariamente se encuentran ajustadas a 1 mol de avance de la reacción, y por ende, los números estequiométricos pueden interpretarse como valores fraccionarios, es decir como cantidades molares. Recuerde que una ecuación química se interpreta tanto molar como molecularmente si su avance de reacción es de 1 mol.
Podemos usar la ley de Hess y tabulaciones de valores de ∆Hfo, para calcular el cambio de entalpía estándar para cualquier reacción para la que conocemos los valores de ∆Hfo para todos los reactivos y productos. En este caso es conveniente tener en cuenta que los signos de las entalpías de los reactivos se invierten, por lo que la ley de Hess debe reescribirse como:

Al simplificar la ecuación anterior puede llegar a ser difícil de manipular, por lo que es más conveniente colocar la identidad de las sustancias para el número estequiométrico y la entalpía sobre cada producto.

Por ende, es fundamental constar con una ecuación química balanceada para masa y carga antes de ejecutar la suma ponderada. En ocasiones debemos calcular el calor desprendido por mol o por gramo de uno de los reactivos empleando los calores de formación estándar.
Enunciado: ¿Cuál es la cantidad más negativa a 25 °C ∆H°f para H2O (l) o ∆H°f para H2O (g)? // Pulse aquí.
Enunciado: Calcule el calor de combustión para 2C2H2(g) + 5O2(g) → 4CO2(g) + 2H2O(l) // Pulse aquí.
Enunciado: Calcule el calor de combustión para C2H4(g) + 3O2(g) → 2CO2(g) + 2H2O(l) // Pulse aquí.
Enunciado: Calcule el calor de combustión para 2H2S(g) + 3O2(g) → 2H2O(l) + 2SO2(g) // Pulse aquí.
Del ejercicio anterior se desprende una pregunta importante con respecto a la simbología estándar de la teoría de termoquímica básica ¿cuál es la relación entre la entalpía estándar de una sustancia y las terapias estándar de formación de una sustancia? en este caso tenemos 2 opciones: (1) que sea en el mismo concepto o (2) que estén relacionadas mediante una constante de proporcionalidad. Para resolver esta pregunta vamos a emplear el enunciado anterior, pero cambiando la pregunta.
Por lo tanto, se hace evidente que los 2 conceptos son diferentes, entonces ¿cuál es la función que relaciona a la entrega estándar de una sustancia con la entalpía estándar de formación?

Eq 12.4. Ley de Hess aplicada a la entalpía por mol de una sustancia.
Eq 12.5. Ley de Hess aplicada a la entalpía por gramo de una sustancia.
Eq 12.6
13. Calor de solución y disolución
13. Calor de solución y disolución
Aunque hasta ahora nos hemos centrado en los efectos de la energía térmica que resultan de las reacciones químicas, muchos procesos físicos, como el derretimiento del hielo o la condensación de un vapor, también implican la absorción o liberación de calor. Los cambios de entalpía también ocurren cuando un soluto se disuelve en un solvente o cuando se diluye una solución. Veamos estos dos procesos físicos relacionados, que involucran calor de solución y calor de dilución.
(13.1) Calor de solución de un sólido iónico
En la gran mayoría de los casos, disolver un soluto en un solvente produce un cambio de calor mensurable. A presión constante, el cambio de calor es igual al cambio de entalpía. El calor de la solución, o entalpía de la solución, ∆Hsln, es el calor generado o absorbido cuando una cierta cantidad de soluto se disuelve en una cierta cantidad de solvente. La cantidad ∆Hsln representa la diferencia entre la entalpía de la solución final y las entalpías de sus componentes originales Hcom (es decir, soluto y solvente) antes de que se mezclen. Por lo tanto,

No se pueden medir ni Hsln ni Hcom, pero su diferencia, ∆Hsln, se puede determinar fácilmente en un calorímetro de presión constante como el de taza de café. Al igual que otros cambios de entalpía, el ∆Hsln.
(a) es positivo para los procesos endotérmicos (que absorben calor) y
(b) negativo para los procesos exotérmicos (que generan calor).
El proceso por el cual se disuelven los sólidos iónicos involucra un proceso a dos pasos,
(a) el primero es el rompimiento de la organización iónica que involucra una energía llamada energía reticular U.
(b) el segundo paso es la integración de los iones desorganizados a la red acuosa por interacciónes entre los monopolos y las moléculas de agua, proceso que requiere de una energía hidratante ∆Hhdr.
Al aplicar la ley de Hess a la energía de la solución, podemos expresarla como la suma de las energías reticular e hidratante.

Tabla 13.1. Entalpías o calores de disolución de algunos compuestos iónicos comunes

(13.2) Calor de disolución de un sólido iónico
En este contexto disolver una solución es agregarle agua, por ende ¿Cómo se comporta energéticamente una solución al disolverse? El calor de dilución es el cambio de calor asociado con el proceso de dilución. Si un determinado proceso de solución es endotérmico y la solución se diluye posteriormente, la misma solución absorberá más calor del entorno. Lo contrario es válido para un proceso de solución exotérmica: se liberará más calor si se agrega más solvente para diluir la solución. Por lo tanto, tenga siempre cuidado al trabajar en un procedimiento de dilución en el laboratorio, algunos solutos pueden comportarse violentamente al solo agregarles agua, siendo los más famosos, los ácidos concentrados como el sulfúrico.
(13.3) Diluyendo el ácido sulfúrico
Diluir ácido sulfúrico concentrado con agua libera una cantidad considerable de calorías en los alrededores. Este proceso es tan exotérmico que nunca debe intentar diluir el ácido concentrado agregándole agua. El calor generado podría hacer que la solución ácida hierva y salpique, y créanme cuando les digo que nadie quiere gotas de ácido sulfúrico volando por un pasillo estrecho de laboratorio lleno de gente. El procedimiento recomendado es agregar el ácido concentrado lentamente al agua (mientras se agita constantemente).
14. Alimentos
14. Alimentos
La mayoría de las reacciones químicas utilizadas para la producción de calor son reacciones de combustión. La energía liberada cuando se quema un gramo de cualquier sustancia es el valor combustible de la sustancia. El valor de combustible de cualquier alimento o combustible se puede medir por calorimetría.
(14.1) La glucosa como fuente de energía
La mayor parte de la energía que nuestro cuerpo necesita proviene de los carbohidratos y las grasas. Los carbohidratos conocidos como almidones se descomponen en los intestinos en glucosa, C6H12O6. La glucosa es soluble en sangre y en el cuerpo humano se la conoce como azúcar en sangre. Es transportada por la sangre a las células donde reacciona con el O2 en una serie de pasos que puede estudiar en los capítulos de glucólisis y respiración celular, produciendo finalmente CO2(g), H2O(l) y energía con una entalpía estándar de -2803 kJ/mol de reacción (o de glucosa, es este caso da lo mismo).
Debido a que los carbohidratos se descomponen rápidamente, su energía se suministra rápidamente al cuerpo. Sin embargo, el cuerpo almacena solo una cantidad muy pequeña de carbohidratos. El valor medio de combustible de los carbohidratos es 17 kJ/g o 4 kcal/g.
(14.2) Las grasas como fuente de energía
Al igual que los carbohidratos, las grasas producen CO2 y H2O cuando se metabolizan. La reacción de la triestearina, C57H110O6, una grasa típica genera una entalpía estándar de 275520 kJ/mol de reacción.
El cuerpo utiliza la energía química de los alimentos para mantener la temperatura corporal, contraer los músculos y construir y reparar tejidos. Cualquier exceso de energía se almacena en forma de grasas. Las grasas son adecuadas para servir como reserva de energía del cuerpo por al menos dos razones: (a) son insolubles en agua, lo que facilita el almacenamiento en el cuerpo, y (b) producen más energía por gramo que las proteínas o los carbohidratos, que las convierte en fuentes de energía compactas. El valor medio de las grasas en el combustible es 38 kJ/g o 9 kcal/g.
(14.3) Midiendo la entalpía de una reacción en un paso o en múltiples pasos
La combustión de carbohidratos y grasas en un calorímetro de bomba genera los mismos productos que cuando se metabolizan en el cuerpo. Aunque evidentemente en el cuerpo las sustancias orgánicas no se oxidan en una combustión, o si no arderíamos espontáneamente, lo que sucede es que la entalpía estándar de una reacción ∆Ho al ser una variable de estado, tiene el mismo valor sin importar que se haga en un solo paso de combustión en un calorímetro de bomba o que se haga en un sinfín de pequeños pasos como ocurre en un proceso metabólico biológico.
(14.4) Proteínas como fuentes de energía
El metabolismo de las proteínas produce menos energía que la combustión en un calorímetro porque los productos son diferentes. Las proteínas contienen nitrógeno, que se libera en el calorímetro de bomba como N2. En el cuerpo, este nitrógeno termina principalmente como urea, (NH2)2CO. El cuerpo utiliza las proteínas principalmente como materiales de construcción para las paredes de los órganos, la piel, el cabello, los músculos, etc. En promedio, el metabolismo de las proteínas produce 17 kJ/g o 14 kcal/g, lo mismo que para los carbohidratos.
(14.5) Tablas de valor de combustible en alimentos
Los valores de combustible para algunos alimentos comunes se muestran en la siguiente tabla:
Tabla 14.1. Composiciones y valores de combustible de algunos alimentos comunes. Tomada de la Química La Ciencia Central (Brown et al., 2017). Tenga en cuenta que los alimentos presentados aquí se toman como mezclas de sustancias con sus propios valores de combustible promedio. Algunos alimentos tienen proporciones muy bajas de algunos nutrientes, por ejemplo, la mantequilla contendrá muy pocos carbohidratos o proteínas en comparación con la proporción en grasa que posee, por tal razón en la tabla cuando eso sucede se coloca un guion.
|

Las etiquetas de los alimentos envasados muestran las cantidades de carbohidratos, grasas y proteínas contenidas en una porción promedio, así como la cantidad de energía suministrada por una porción. La cantidad de energía que nuestro cuerpo necesita varía considerablemente, dependiendo de factores como el peso, la edad y la actividad muscular. Se requieren aproximadamente 100 kJ por kilogramo de masa corporal por día para mantener el funcionamiento del cuerpo a un nivel mínimo. Una persona promedio de 70 kg (154 libras) gasta alrededor de 800 kJ/h cuando realiza un trabajo ligero, y la actividad intensa a menudo requiere 2000 kJ/h o más. Cuando el valor de combustible, o el contenido calórico, de los alimentos que ingerimos excede la energía que gastamos, nuestro cuerpo almacena el excedente en forma de grasa.
(14.6) Calculando el valor de combustible
De una sustancia pura
El valor de la combustión va a ser igual al cociente que ocurre entre la entalpía estándar de la reacción sobre la masa del compuesto clave.

De una mezcla
Como vimos anteriormente, los alimentos y algunos tipos de combustibles son en realidad mezclas de sustancias y por lo tanto, se hace necesario pode calcular el valor de combustible total.

Valor de combustible total en términos de la fracción de masas de cada componente y su respectivo valor de combustible.
De una porción
El valor de combustible de una porción va a tener unidades de energía únicamente ya que ésta está dada para una masa arbitraria de la sustancia. Dado lo anterior denominaremos a esta variable como Portion Fuel Value (PFV).
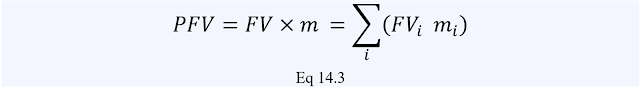
Variables de consumo y porciones
La energía de los alimentos se consume esencialmente por dos variables, el tiempo t o la distancia recorrida x. ¿Cuantas porciones deben consumirse para proporcionar la energía necesaria para mantener un determinado rendimiento en un tiempo t o una distancia x?

Porciones de consumo en términos de una variable de estado como desplazamiento, tiempo, presión, fuerza, una constante lineal y un valor de combustible por porción.
15. Combustibles fósiles y otras fuentes de energía
15. Combustibles fósiles y otras fuentes de energía
Durante la combustión completa de los combustibles, el carbono se convierte en CO2 y el hidrógeno se convierte en H2O, los cuales tienen grandes entalpías de formación negativas. En consecuencia, cuanto mayor es el porcentaje de carbono e hidrógeno en un combustible, mayor es su valor de combustible. En la Tabla 15.1, por ejemplo, compare las composiciones y los valores de combustible del carbón bituminoso y la madera de pino. El carbón tiene un valor combustible más alto debido a su mayor contenido de carbono.
Tabla 15.1. Valores de combustible y composiciones de algunos combustibles comunes. Tomada de la Química La Ciencia Central (T. L. Brown et al., 2017).

En 2011, Estados Unidos consumió 1.03 x 1017 kJ de energía siendo suministrada por carbón 22.6%, gas natural 24.0%, petróleo 37.6%, nuclear 8.5% y fuentes renovables 7.4%. Este valor corresponde a un consumo medio diario de energía por persona de 9.3 x 105 kJ, aproximadamente 100 veces mayor que las necesidades de energía alimentaria per cápita.
(15.1) Combustibles fósiles
El carbón, el petróleo y el gas natural, que son las principales fuentes de energía del mundo, se conocen como combustibles fósiles. Todos se han formado durante millones de años a partir de la descomposición de plantas y animales marinos y se están agotando mucho más rápidamente de lo que se están formando.
Gas natural
El gas natural se compone de hidrocarburos gaseosos, compuestos de hidrógeno y carbono. Contiene principalmente metano CH4, con pequeñas cantidades de etano C2H6, propano C3H8 y butano C4H10. El gas natural se quema con muchos menos subproductos y produce menos CO2 que el petróleo o el carbón.
Petróleo
El petróleo es un líquido compuesto por cientos de compuestos, la mayoría de los cuales son hidrocarburos, y el resto son principalmente compuestos orgánicos que contienen azufre, nitrógeno u oxígeno.
Carbón mineral
El carbón, que es sólido, contiene hidrocarburos de alto peso molecular, así como compuestos que contienen azufre, oxígeno o nitrógeno. El carbón es el combustible fósil más abundante; Se prevé que las reservas actuales duren más de 100 años a las tasas de consumo actuales. Sin embargo, el uso de carbón presenta varios problemas. El carbón es una mezcla compleja de sustancias y contiene componentes que contaminan el aire. Cuando el carbón se quema, el azufre que contiene se convierte principalmente en dióxido de azufre, SO2, un contaminante molesto del aire. Debido a que el carbón es un sólido, la recuperación de sus depósitos subterráneos es costosa y, a menudo, peligrosa tanto por retos tecnológicos como de geopolítica. Además, los depósitos de carbón no siempre están cerca de lugares de uso de alta energía, por lo que a menudo hay costos de envío sustanciales.
Gas de invernadero
Los combustibles fósiles liberan energía en las reacciones de combustión, que idealmente producen solo CO2 y H2O. La producción de CO2 se ha convertido en un tema importante que involucra a la ciencia y las políticas públicas debido a la preocupación de que las crecientes concentraciones de CO2 atmosférico estén causando cambios climáticos globales.
(15.2) Otras fuentes de energía
Nuclear
La energía nuclear es la energía liberada en la fisión (división) o la fusión (combinación) de núcleos atómicos. La energía nuclear basada en la fisión nuclear se utiliza actualmente para producir aproximadamente el 21% de la energía eléctrica en los Estados Unidos y representa aproximadamente el 8.5% de la producción total de energía de los EE. UU. La energía nuclear está, en principio, libre de las emisiones contaminantes que son un problema importante con los combustibles fósiles. Sin embargo, las plantas de energía nuclear producen desechos radiactivos, por lo que su uso ha sido controvertido.
Renovables
Los combustibles fósiles y la energía nuclear son fuentes de energía no renovables; son recursos limitados que consumimos a un ritmo mucho mayor de lo que pueden regenerarse. Con el tiempo, estos combustibles se gastarán, aunque las estimaciones varían mucho en cuanto a cuándo ocurrirá esto. Debido a que las fuentes de energía no renovables eventualmente se agotarán, y con agotar nos referimos a que con el tiempo serán demasiado costosas para obtener y comerciar, se está llevando a cabo una gran cantidad de investigación sobre las fuentes de energía renovables, fuentes que son esencialmente inagotables.
Las fuentes de energía renovable incluyen la energía solar del Sol, la energía eólica aprovechada por los molinos de viento, la energía geotérmica del calor almacenado dentro de la Tierra, la energía hidroeléctrica de los ríos que fluyen y la energía de biomasa de los cultivos y los desechos biológicos. Actualmente, las fuentes renovables proporcionan alrededor del 7.4% del consumo anual de energía de los EE. UU., Siendo las fuentes hidroeléctricas y de biomasa las principales contribuyentes.
Satisfacer nuestras necesidades energéticas futuras dependerá del desarrollo de tecnología para aprovechar la energía solar con mayor eficiencia. La energía solar es la fuente de energía más grande del mundo. En un día despejado, aproximadamente 1 kJ de energía solar llega a cada metro cuadrado de la superficie de la Tierra cada segundo. La energía solar promedio que cae en solo el 0.1% de la superficie terrestre de los EE. UU. Es equivalente a toda la energía que usa actualmente esta nación. Aprovechar esta energía es difícil porque está diluida (es decir, distribuida en un área amplia) y varía con la hora del día y las condiciones climáticas. El uso eficaz de la energía solar dependerá del desarrollo de algún medio de almacenamiento y distribución. Es casi seguro que cualquier medio práctico para hacer esto involucrará un proceso químico endotérmico que luego se puede revertir para liberar calor. Una de esas reacciones es: CH4(g) + H2O(g) + calor → CO(g) + 3 H2(g). Esta reacción avanza hacia adelante a altas temperaturas, que se pueden obtener en un horno solar. El CO y el H2 formados en la reacción podrían almacenarse y dejarse reaccionar más tarde de manera inversa empleando una chispa, con el calor liberado para un trabajo útil.
Las plantas utilizan la energía solar en la fotosíntesis, la reacción en la que la energía de la luz solar se utiliza para convertir CO2 y H2O en carbohidratos y O2: 6 CO2(g) + 6 H2O(l) + luz → C6H12O6(s) + 6 O2(g). La fotosíntesis es una parte importante del ecosistema de la Tierra porque repone el O2 atmosférico, produce una molécula rica en energía que puede usarse como combustible y consume algo de CO2 atmosférico. Quizás la forma más directa de utilizar la energía del Sol es convertirla directamente en electricidad en dispositivos fotovoltaicos, o células solares. Las eficiencias de estos dispositivos se han incrementado drásticamente durante los últimos años. Los avances tecnológicos han llevado a paneles solares que duran más y producen electricidad con mayor eficiencia a un costo unitario cada vez menor. De hecho, el futuro de la energía solar es, como el propio Sol, muy brillante.
16. La energía del futuro
16. La energía del futuro
La energía es un factor que determina la economía, la infraestructura, el transporte y el nivel de vida de un país. El problema que se enfrenta a nivel mundial es la disparidad entre el consumo y la disponibilidad de energía (lo cual no sólo involucra la cantidad de éstas sino los lugares donde se produce y sus condiciones socioculturales, lo cual normalmente desencadena en conflictos bélicos) (Manoharan et al., 2019).
Todas las naciones actualmente dependen de los combustibles fósiles para la producción de energía, y estos combustibles fósiles no son fuentes sostenibles. Para satisfacer las demandas de energía de la población mundial que crece más rápidamente, es esencial actualizarse a una fuente de energía alternativa y sostenible que no afecte negativamente al medio ambiente (Manoharan et al., 2019).
En las últimas décadas, Estados Unidos ha puesto énfasis en el impacto ambiental del sector del transporte y en la reducción de la dependencia del petróleo. El agotamiento de los combustibles convencionales no renovables es uno de los principales problemas del escenario energético moderno y geopolítico, lo que hace que el estado de la industria energética sea insostenible, y también causa problemas ambientales como el efecto invernadero. Hoy en día, la proporción del uso de combustibles fósiles sigue siendo alta y se prevé que representará aproximadamente el 75 % de la producción de energía en 2050 (Manoharan et al., 2019).
En general, el escenario energético actual tiene muchas desventajas. Sin embargo, existen muchas fuentes de energía sostenibles, y si su uso aumenta, el escenario será mucho más optimista para las generaciones futuras. Los ambientalistas pronostican que el peor de los escenarios del calentamiento global y sus efectos no se alcanzará debido a diferentes iniciativas. En las últimas dos décadas, los vehículos se han vuelto más eficientes en combustible y los vehículos eléctricos híbridos se están volviendo más comunes. Una de las alternativas de más rápido crecimiento en los vehículos es la electricidad. Al igual que con las fuentes de energía convencionales como el petróleo y el carbón, la electricidad no es una fuente de energía primaria.
Una batería totalmente cargada es un portador de energía. Los vehículos eléctricos de batería (BEV) son muy eficientes para convertir la energía de la red en fuerza de tracción, y pueden recuperar energía durante la conducción utilizando el frenado regenerativo. Un inconveniente importante de los BEV es que, por lo general, tienen una autonomía limitada debido al tamaño y el costo de las baterías necesarias para los requisitos de potencia y energía del vehículo. El “repostaje” de los sistemas de baterías también puede llevar varias horas, en lugar de unos pocos minutos con un vehículo convencional (CV). Para utilizar las ventajas de los vehículos eléctricos y convencionales y cerrar la brecha entre CV y BEV, se considera una alternativa (Manoharan et al., 2019).
El hidrógeno es un portador de energía química que tiene la capacidad de producir electricidad hasta 39.39 kWh/kg, lo que supera la densidad energética de la mayoría de las baterías (Manoharan et al., 2019). De hecho, en la sección anterior se observa a los valores de combustible, observará que el hidrógeno posee un valor de combustible de 142 kJ/g mientras que la gasolina sólo alcanza 48 kJ/g, lo cual implica necesariamente que, como portador de energía, el hidrógeno puede almacenar energía más eficientemente que cualquier combustible conocido hasta el momento.
Una celda de combustible (FC) tiene una analogía directa con un motor de combustión interna (ICE). Un ICE convierte la energía química almacenada en el combustible suministrado al motor para producir energía mecánica rotacional. La energía de rotación producida se usa para impulsar un vehículo o se concentra a través de un generador y se convierte en energía eléctrica. Un FC actúa de la misma manera que un ICE en el sentido de que la energía química se convierte directamente en energía eléctrica en el FC, pero en un proceso respetuoso con el medio ambiente pues el gas producido es vapor de agua en lugar de dióxido de carbono. A diferencia de una batería que se agota mientras se usa para alimentar componentes eléctricos, los motores de combustión interna y las celdas de combustible actúan como fuentes de energía continuamente operativas siempre que se les suministre combustible. Por lo tanto, se proyecta que la celda de combustible de hidrógeno pueda superar las desventajas de los BEV, convirtiendo al hidrógeno en el combustible de transporte del futuro (Manoharan et al., 2019).
(16.1) Combustible de hidrógeno
El hidrógeno es la forma más simple de todas las moléculas; tiene el contenido de energía más bajo por volumen, pero tiene el contenido de energía más alto de cualquier combustible por peso. Esta aparente paradoja se debe a que el hidrógeno es muy poco denso, y por lo tanto se puede almacenar mucha cantidad química de él, es decir muchos moles, en una cantidad de espacio muy pequeña (Manoharan et al., 2019).
Está disponible en la atmósfera como gas y en el agua como líquido. Debido al alto contenido energético del hidrógeno, se emplea como combustible en aplicaciones como celdas de combustible y cohetes. El hidrógeno genera cero emisiones nocivas, que es uno de los inconvenientes más importantes de los combustibles fósiles, y el poder calorífico del hidrógeno es tres veces mayor que el del petróleo (Manoharan et al., 2019).
El principal problema del hidrógeno es que no viene prefabricado por un proceso natural que lo genere en enormes cantidades, como sí sucede con la gasolina, que se puede refinar fácilmente desde el petróleo. Con el hidrógeno la cosa es más complicada pues debemos meter energía a otras sustancias para extraerlo. Hay un alto costo de producción involucrado con el hidrógeno, ya que es un combustible hecho por el hombre, que cuesta aproximadamente tres veces más que la refinación de petróleo (Manoharan et al., 2019).
Se dedican cantidades sustanciales de investigación a crear una forma eficiente y sostenible de producir hidrógeno y aplicaciones para el hidrógeno en los motores de transporte. Los fabricantes de automóviles como Honda, Toyota y Hyundai han comenzado a fabricar vehículos de pila de combustible (FCV) con hidrógeno como combustible. Estos FCV están disponibles actualmente en América del Norte, Asia y Europa, y han sido adquiridos principalmente por los primeros usuarios. Los consumidores actuales, los primeros en adoptar, son principalmente personas con un alto nivel educativo, familias de altos ingresos, aquellos con unidades familiares más grandes, aquellos que están dispuestos a cambiar su estilo de vida y aquellos con otros atributos similares (Manoharan et al., 2019).
Hasta junio de 2018, se han vendido más de 6500 FCV a los consumidores. California es el mercado líder para FCV, con casi 3000 FCV entregados allí de los 5233 vehículos vendidos a nivel mundial debido a que el estado alberga la red más grande de estaciones de servicio de hidrógeno en el mundo y los fabricantes de automóviles venden los vehículos allí. En la actualidad, varios fabricantes de automóviles están promocionando los FCV entre los consumidores, que a menudo se comparan con los BEV. Tanto los BEV como los FCV ofrecen cero emisiones del tubo de escape, la capacidad de alimentarse con fuentes de energía renovables y sostenibles y utilizar motores eléctricos. Las diferencias más notables entre los FCV y los BEV son la autonomía de conducción y el estilo de repostaje. Los FCV tienen un rango de manejo de más de 300 millas y se pueden repostar en menos de 10 minutos en una estación de reabastecimiento de hidrógeno, que es más comparable a un vehículo tradicional ICE impulsado por combustibles fósiles (Manoharan et al., 2019).
El hidrógeno tiene un mayor potencial para su uso como combustible en el futuro. Se estima que, para el año 2030, el costo de las celdas de combustible será competitivo con los ICE en función de las mejoras tecnológicas que se están realizando y la mayor disponibilidad [14]. Uno de los principales obstáculos que enfrenta el uso masivo de hidrógeno es un almacenamiento más eficiente. Debido a la baja densidad del hidrógeno, no se puede almacenar tan fácilmente como los combustibles fósiles tradicionales, eso se debe a que si abres descuidadamente un contenedor con hidrógeno el gas se va a escapar, y si hay una leve chispa, te tendrán que recoger con espátula del piso después de ser calcinado por la explosión (Manoharan et al., 2019).
El hidrógeno requiere compresión, enfriamiento o una combinación de ellos. El método más favorable de almacenamiento de hidrógeno es la contención física, específicamente en tanques comprimidos, porque están fácilmente disponibles. Se utilizan principalmente todos los compuestos (Tipo IV) y, a veces, se utilizan compuestos revestidos de metal (Tipo III). El tiempo de llenado de estos tanques es competitivo con el de los combustibles fósiles cuando el hidrógeno se enfría previamente (Manoharan et al., 2019).
El costo es el principal inconveniente para el uso a gran escala de tanques de hidrógeno comprimido (CH2), debido a que el material y el montaje son caros. Otro revés potencial es la preocupación del público por el uso de tanques de almacenamiento de alta presión (70 MPa) en los vehículos. Una alternativa a los tanques de CH2 tradicionales que aún se está investigando es un tanque con un esqueleto interno, que es un diseño complejo de puntales en tensión dentro del tanque para resistir las fuerzas del gas comprimido. El almacenamiento de hidrógeno líquido (LH2) ha mejorado significativamente, logrando la mejor masa específica (15 %) de cualquier otro sistema de almacenamiento de hidrógeno automotriz. La eficiencia energética disminuye cuando se utiliza hidrógeno líquido. La ebullición es un área que necesita mejoras antes de que los sistemas LH2 puedan ser ampliamente aceptados. Un diseño alternativo prometedor es un tanque criocomprimido en el que el hidrógeno está altamente comprimido a temperaturas criogénicas. Se deben realizar más estudios sobre este método para determinar la durabilidad a largo plazo y la aceptación pública del sistema.
Los sistemas de almacenamiento de hidruros requieren una investigación y un progreso sustanciales para cumplir con los requisitos para el uso a gran escala. El hidruro mejor estudiado es NaAlH4, pero no tiene la capacidad necesaria para su aplicación. En base a los resultados de los pocos estudios existentes, se indica que se podrían construir tanques sin elementos internos de transferencia de calor basados en el moderado calor de absorción del hidrógeno en las superficies (Manoharan et al., 2019).
Aunque la presencia de hidrógeno es abundante en la atmósfera, no se encuentra en la forma más pura. El hidrógeno se puede extraer del agua, combustible de hidrocarburo, sulfuro de hidrógeno y otros elementos químicos. La energía que se utiliza para producir hidrógeno a partir de sus elementos asociados requiere energía externa, como energía térmica, eléctrica, fotónica y bioquímica. El amoníaco, tiene un alto porcentaje de hidrógeno en su interior y se propuso como combustible para motores de combustión interna a través de la descomposición a bordo en hidrógeno y nitrógeno (Manoharan et al., 2019).
El hidrógeno se puede extraer de fuentes de energía renovables o no renovables. La producción de hidrógeno a partir de energías renovables siempre es respetuosa con el medio ambiente, mientras que el hidrógeno producido a partir de fuentes no renovables emite gases de efecto invernadero (GEI). La producción de hidrógeno a partir de la biomasa residual mediante reacciones electroquímicas es libre de GEI y no requiere una gran cantidad de energía ni altos costos de producción. Las posibles materias primas de biomasa son los residuos de pan, el aserrín de ciprés y la paja de arroz. Similar a estos, el periódico podría usarse para producir hidrógeno por electrólisis directa. El papel periódico está compuesto por 69.2 % de celulosa y 11.8 % de lignina, y se descompone en monosacáridos y disacáridos, así como en cetoácido alifático en el solvente H3PO4 en condiciones similares a las de la electrólisis. El hidrógeno también puede generarse mediante la electrólisis del metano humidificado (Manoharan et al., 2019).
(16.2) Una fuente de energía eficiente
Como se ha recalcado anteriormente, el hidrógeno es sólo un portador de energía, por lo que en últimos hay que extraerla de algún lado en donde ya esté almacenada previamente en cantidades industriales, y además sea económico y seguro de extraer. El hecho de que aún estemos anclados al sistema de combustibles fósiles revela que aún no se tiene una respuesta clara para estos inconvenientes, sin embargo, muchos en la comunidad científica esperamos que la fusión nuclear sea la respuesta final.
Hay un dicho que dice que la fusión nuclear siempre está a dos décadas de distancia, y revistas científicas como Nuclear Fusion de la Agencia Internacional de Energía Atómica han relatado la larga “y lenta” evolución de la energía de fusión. Dicho esto, en el año del 75 aniversario de Hiroshima y Nagasaki, ITER, el experimento de fusión más grande del mundo, comenzó su ensamblaje de máquinas, colocando así la energía de fusión dentro de la planificación militar de largo alcance. El líder de al menos una gran potencia mundial ahora enfatiza públicamente que la energía de fusión debe usarse con fines pacíficos, una admisión tácita de la alternativa: el uso de la fusión como arma, que paradójicamente fue la primera en alcanzarse durante la guerra fría entre Estados Unidos y la Unión Soviética (Carayannis, Draper, & Bhaneja, 2021).
La fusión de hidrógeno es un método ideal para generar energía. El combustible de deuterio (el cual a su vez es un isótopo del hidrógeno, pero en lugar de generar una reacción química de combustión, en este caso estamos hablando de una reacción nuclear de fusión) está fácilmente disponible en el agua de mar y el único subproducto es el helio. Al igual que una planta de gas, carbón o fisión, una planta de fusión proporcionará energía de carga base altamente concentrada durante todo el día. Sin embargo, la fusión no produce emisiones de gases de efecto invernadero ni desechos radiactivos de larga duración. El riesgo de accidentes con una planta de fusión es muy limitado: si se pierde la contención, la reacción de fusión simplemente se detiene.
La energía de fusión está más cerca de lo que mucha gente cree. Podría proporcionar una fuente de electricidad libre de carbono para la red, desempeñando un papel clave a medida que EE. UU. y otras naciones descarbonizan su infraestructura de generación de energía. En ese escenario la energía producida por un reactor de fusión puede emplearse para extraer hidrógeno puro de fuentes baratas como el agua de mar, en ese punto el hidrógeno puede transportarse a la red de suministro de energía para los automóviles y para la industria de una manera homóloga a cómo se hace con el combustible fósil. A diferencia del combustible fósil, el agua de mar se encuentra disponible fácilmente para la mayoría de las potencias económicas globales, por lo que la importancia de países que poseen fuentes de combustibles fósiles a nivel geopolítico disminuiría drásticamente, lo cual implicaría una caída abrupta en los precios del petróleo.
Dos informes recientes publicados por la comunidad de fusión exponen las formas en que EE. UU. puede llegar allí. En diciembre, el Comité Asesor de Ciencias de la Energía de Fusión del Departamento de Energía de EE. UU. publicó un informe que establece un plan estratégico para la investigación de la ciencia del plasma y la energía de fusión durante la próxima década. Pide el desarrollo y la construcción de una planta piloto de fusión para 2040. En febrero de 2021, las Academias Nacionales de Ciencias, Ingeniería y Medicina (NASEM) publicaron un informe complementario que pedía acciones agresivas para construir una planta de energía piloto. El informe de NASEM propone un diseño para 2028 y una planta piloto de fusión en la línea de tiempo 2035-2040.
“El objetivo de trabajar a partir de esta línea de tiempo fue delinear lo que se necesitaría para tener un impacto en la transición hacia la reducción de las emisiones de carbono para mediados de siglo. Muchas inversiones y actividades esenciales tendrían que comenzar ahora para cumplir con ese cronograma”, dice Kathy McCarthy, Directora de la Oficina de Proyectos ITER de EE. UU. en el Laboratorio Nacional de Oak Ridge. “La experiencia que estamos obteniendo de ITER en ingeniería integrada a escala de reactor es invaluable para realizar un camino viable y práctico hacia la energía de fusión”.
Referencias bibliográficas
Referencias bibliográficas
Bacon, F. (1878). Novum organum. Clarendon press.
Baeza-Baeza, J. J., & García-Alvarez-Coque, M. C. (2014). Extent of Reaction Balances: A Convenient Tool to Study Chemical Equilibria. World J. Chem. Educ, 2, 54–58. Retrieved from https://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.1015.5804&rep=rep1&type=pdf
Bernoulli, D. (1738). Hydrodynamica, sive de Viribus et Motibus Fluidorum Commentarii (hydrodynamics, or commentaries on the forces and motions of fluids) Basel.
Boyle, R. (1911). The Sceptical Chymist: The Classic 1661 Text. Courier Dover Publications.
Brown, A. (1999). Modern Thermodynamics: From Heat engines to Dissipative Structures. Proceedings of the Institution of Mechanical Engineers, 213(1), 63.
Brown, L., & Holme, T. (2013). Introduction to Chemistry.
Brown, T. L., LeMay, H. E. J., Bursten, B. E., Murphy, C. J., Woodward, P. M., & Stoltzfus, M. W. (2017). Chemistry, the central science (13th ed.). Boston: Pearson.
Brown, T. L., LeMay, H. E. J., Bursten, B. E., Murphy, C. J., Woodward, P., & Stoltzfus, M. W. (2015). Chemistry the Central Science.
Burdge, J., & Overby, J. (2018). Chemistry: atoms first (3rd ed.). McGraw-Hill Education.
Buyse, F. (2020). Heat in Renaissance Philosophy.
Canagaratna, S. G. (2000). The use of extent of reaction in introductory courses. Journal of Chemical Education, 77(1), 52. Retrieved from https://pubs.acs.org/doi/abs/10.1021/ed077p52
Carayannis, E. G., Draper, J., & Bhaneja, B. (2021). Towards fusion energy in the Industry 5.0 and Society 5.0 context: Call for a global commission for urgent action on fusion energy. Journal of the Knowledge Economy, 12(4), 1891–1904.
Carnot, S. (1824). Reflections on the motive power of fire, and on machines fitted to develop that power. Paris: Bachelier, 108, 1824.
Cercignani, C. (2001). The Rise of Statistical Mechanics. In Chance in Physics (pp. 25–38). Springer.
Chalmers, A. (2009). Atomism: Science or Philosophy? In The Scientist’s Atom and the Philosopher’s Stone (pp. 1–17). Springer.
Chang, R. (2006). Chang’s “General Chemistry - Essential Concepts” (4th ed.). McGraw-Hill New York.
Chang, R. (2010). Chemistry (10th ed.). McGraw-Hill New York.
Chang, R., & Overby, J. (2011). General Chemistry,Th e Essential Concepts (11th ed.). McGraw-Hill New York.
Clausius, R. (1857a). Ueber die Art der Bewegung, welche wir Wärme nennen. Annalen Der Physik, 176(3), 353–380.
Clausius, R. (1857b). XI. On the nature of the motion which we call heat. The London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science, 14(91), 108–127.
Croce, A. E. (2002). The application of the concept of extent of reaction. Journal of Chemical Education, 79(4), 506.
De Donder, T., & Van Rysselberghe, P. (1936). Thermodynamic theory of affinity (Vol. 1). Stanford university press.
Fox, R. (2006). What was ‘crucial’about Rumford’s experiments on the nature of heat? Atti Del XI Convegno Nazionale Di Storia e Fondamenti Della Chimica, Roma, Accademia Nazionale Delle Scienze Detta Dei XL, 415–425.
García-García, J. L. (2021). Hacia un equilibrio químico verdaderamente analítico. Educación Química, 32(1), 133–146. Retrieved from http://revistas.unam.mx/index.php/req/article/view/75075
García García, J. L. (2020). El algebra de la estequiometría. Educación Química, 31(1), 138–150.
Garst, J. F. (1974). The extent of reaction as a unifying basis for stoichiometry in elementary chemistry. Journal of Chemical Education, 51(3), 194. Retrieved from https://files.eric.ed.gov/fulltext/EJ1179603.pdf
Gibbs, J. W. (1878). On the equilibrium of heterogeneous substances. American Journal of Science, 3(96), 441–458.
Griffiths, J. G. (1955). The Orders of Gods in Greece and Egypt (According to Herodotus). The Journal of Hellenic Studies, 75, 21–23.
Hanyak Jr, M. E. (2014). Extent of Reaction or Events of Reaction?
IUPAC, McNaught, A. D., & Wilkinson, A. (2019). extent of reaction, ξ. In Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”) (2nd ed.). Blackwell Scientific Publications, Oxford. Retrieved from https://goldbook.iupac.org/terms/view/E02283
Joule, J. P. (1854). James Prescott Joule. Children, 1000(1847).
Kenny, A. (1964). Vacuum Theory and Technique Greek Science. Transactions of the Newcomen Society, 37(1), 47–56.
Lecoustre, V. R. (2009). Numerical investigations of gaseous spherical diffusion flames. University of Maryland, College Park.
Manoharan, Y., Hosseini, S. E., Butler, B., Alzhahrani, H., Senior, B. T. F., Ashuri, T., & Krohn, J. (2019). Hydrogen fuel cell vehicles; current status and future prospect. Applied Sciences, 9(11), 2296.
Mendelssohn, K. (1977). Quest for absolute zero: the meaning of low temperature physics.
Meschel, S. V. (2020). A brief history of heat measurements by calorimetry with emphasis on the thermochemistry of metallic and metal-nonmetal compounds. Calphad, 68, 101714.
Middleton, W. E. K. (1963). The place of Torricelli in the history of the barometer. Isis, 54(1), 11–28.
Moretti, G. (2015). The “extent of reaction”: a powerful concept to study chemical transformations at the first-year general chemistry courses. Foundations of Chemistry, 17(2), 107–115.
Mousavi, A. (2018). Stoichiometry of equations through the Inverse de Donder relation. Chemistry Teacher International, 1(1). Retrieved from https://www.degruyter.com/document/doi/10.1515/cti-2018-0006/html
Newburgh, R., & Leff, H. S. (2011). The Mayer-Joule principle: The foundation of the first law of thermodynamics. The Physics Teacher, 49(8), 484–487.
Newell, S. B. (1980). Chemistry: an introduction. Little Brown and Company.
Onorato, P., Malgieri, M., Polesello, M., Salmoiraghi, A., & Oss, S. (2019). From chance to the physical laws: Toy models to connect the microscopic and macroscopic understanding of physical phenomena. Il Nuovo Cimento C, 42(5), 1–13.
Redhead, P. A. (1999). The ultimate vacuum. Vacuum, 53(1), 137–149.
Siddiqui, N. (2018). PERSPECTIVE OF ‘UNṢUR-E-NĀR (FIRE AS AN ELEMENT) AND ENERGY IN UNANI MEDICINE.
Styer, D. (2019). Entropy as disorder: History of a misconception. The Physics Teacher, 57(7), 454–458.
Taylor, F. S. (1942). The origin of the thermometer. Annals of Science, 5(2), 129–156.
West, J. B. (2013). Torricelli and the ocean of air: the first measurement of barometric pressure. Physiology, 28(2), 66–73.
Wikipedia, S. (2019). Extent of reaction. Retrieved January 31, 2020, from https://en.wikipedia.org/wiki/Extent_of_reaction#cite_note-2
Xue, T.-W., & Guo, Z.-Y. (2019). What is the real Clausius statement of the second law of thermodynamics? Entropy, 21(10), 926.
E. Ejercicios resueltos
E. Ejercicios resueltos
👉 Demostraciones <Ejercicios analíticos para demostrar las respuestas analíticas o algoritmos de solución> (Pulse aquí)
👉 Ejemplos <Ejercicios Propios de la página> (Pulse aquí)
👉 Química general de Chang <Libro de texto de Raymond Chang> (Pulse aquí)
👉 Química la ciencia central <Libro de texto de Theodore E. Brown> (Pulse aquí)
👉 LibreChem <página de internet> (Pulseaquí)
Demostraciones
1. Hallar una función que permite calcular el cambio de energía de un cilindro de émbolo móvil si este permite el flujo de calor en términos del cambio de entalpía y el cambio de volumen a presión constante.
5. Hallar la ecuación para convertir entre entalpía estándar J/mol y entalpía específica J/g
Ejemplos
Química General de Chang
(Química-Chang10-ejemplo-6.1) Cierto gas se expande en volumen de 2.0 L a 6.0 L a temperatura constante. Calcule el trabajo realizado por el gas si se expande (a) contra el vacío y (b) contra una presión constante de 1.2 atm.
(Química-Chang10-practica-6.1) Un gas se expande de 264 mL a 971 mL a temperatura constante. Calcule el trabajo realizado (en julios) por el gas si se expande (a) contra el vacío y (b) contra una presión constante de 4.00 atm.
(Química-Chang10-ejemplo-6.2) El trabajo realizado cuando se comprime un gas en un cilindro de embolo móvil es 462 J. Durante este proceso, hay una transferencia de calor de 128 J desde el gas al entorno. Calcula el cambio de energía para este proceso.
(Química-Chang10-practica-6.2) Un gas se expande y realiza un trabajo P-V en el entorno igual a 279 J. Al mismo tiempo, absorbe 216 J de calor del entorno. ¿Cuál es el cambio de energía del sistema?
(Química-Chang10-ejemplo-6.3) Dada la ecuación termoquímica 2SO2 (g) + O2 (g) → 2SO3 (g) ΔH = -198.2 kJ / mol calcule el calor desprendido cuando 87.9 g de SO2 (masa molar = 64.07 g / mol) se convierte en SO3
(Química-Chang10-practica-6.3) Calcule el calor desprendido cuando 266 g de fósforo blanco (P4) arden en el aire de acuerdo con la ecuación P4(s) + 5O2(g) → P4O10(s) ΔH = -3013 kJ/mol
Química-Chang10-ejemplo-6.4) Calcule el cambio en la energía interna cuando 2 moles de CO se convierten en 2 moles de CO2 a 1 atm y 25 °C: 2CO(g) + O2 (g) → 2CO2 (g) ΔH = -566.0 kJ/mol
(Química-Chang10-practica-6.4) ¿Cuál es cambio en la energía interna para la formación de 1.00 mol de CO a 1.00 atm y 25°C? C (grafito) + ½ O2 (g) → CO (g) ΔH°= -110.5 kJ/mol
(Química-Chang10-ejemplo-6.5) Se calienta una muestra de agua de 466 g de 8.50 °C a 74.60 °C. Calcula la cantidad de calor absorbido (en kilojulios) por el agua.
(Química-Chang10-practica-6.5) Una barra de hierro de 869 g de masa se enfría de 94 °C a 5 °C. Calcula el calor liberado (en kilojulios) por el metal.
(Química-Chang10-ejemplo-6B) La combustión de cierta masa de ácido benzoico, C6H5COOH, en un calorímetro de bomba produce 26.42 kJ de calor con aumento en la temperatura de 4.673 °C. Determine la capacidad calorífica estándar de dicho calorímetro.
(Química-Chang10-practica-6.6) Se quemó 1.922 g de metanol (CH3OH) en un calorímetro de bomba. En consecuencia, la temperatura del agua aumentó 4.20 °C. Si la capacidad calorífica de la bomba más agua fue de 10.4 kJ/°C, calcule el calor molar de combustión del metanol.
(Química-Chang10-practica-6.9) Calcule la entalpía estándar de formación del disulfuro de carbono (CS2). (a) C (grafito) + O2 ( g) → CO2 (g) ∆H0 = -393.5 kJ/mol (b) S (rómbico) + O2 (g) → SO2 (g) ∆H0 = -296.4 kJ / mol (c) CS2 (l) + 3O2 (g) → CO2 (g) + 2SO2 (g) ∆H0 = -1073.6 kJ / mol.
(Química-Chang10-practica-6.10) El benceno (C6H6) se quema en el aire para producir dióxido de carbono y agua líquida. Calcule el calor liberado (en kilojulios) por gramo del compuesto que reaccionó con oxígeno. La entalpía estándar de formación de benceno es 49.04 kJ/mol
(Química-Chang10-ejercicio-6.15) Una muestra de nitrógeno gaseoso se expande en volumen de 1.6 L a 5.4 L a temperatura constante. Calcule el trabajo realizado en julios si el gas se expande (a) contra el vacío, (b) contra una presión constante de 0.80 atm y (c) contra una presión constante de 3.7 atm.
(Química-Chang10-ejercicio-6.16) Un gas se expande en volumen de 26.7 mL a 89.3 mL a temperatura constante. Calcule el trabajo realizado (en julios) si el gas se expande (a) contra el vacío, (b) contra una presión constante de 1.5 atm y (c) contra una presión constante de 2.8 atm.
(Química-Chang10-ejercicio-6.17) Un gas se expande y hace un trabajo P-V en el entorno igual a 325 J. Al mismo tiempo, absorbe 127 J de calor del entorno. Calcula el cambio de energía del gas.
(Química-Chang10-ejercicio-6.18) El trabajo realizado para comprimir un gas es 74 J. Como resultado, se emiten 26 J de calor al entorno. Calcula el cambio de energía del gas.
(Química-Chang10-ejercicio-6.19) Calcule el trabajo realizado cuando 50.0 g de estaño se disuelven en exceso de ácido a 1.00 atm y 25 ° C: Sn (s) + 2H+ (aq) → Sn2+ (aq) + H2 (g) Suponga el comportamiento del gas ideal.
(Química-Chang10-ejercicio-6.20) Calcule el trabajo realizado en julios cuando 1.0 mol de agua se vaporiza a 1.0 atm y 100 °C. Suponga que el volumen de agua líquida es insignificante en comparación con el del vapor a 100 ° C y el comportamiento del gas ideal.
(Química-Chang10-ejercicio-6.25) El primer paso en la recuperación industrial de zinc a partir del mineral de sulfuro de zinc es el tostado, es decir, la conversión de ZnS en ZnO por calentamiento: 2ZnS (s) + 3O2 (g) → 2ZnO (s) + 2SO2 (g) ∆H0 = -879 kJ/mol. Calcule el calor desprendido (en kJ) por gramo de ZnS.
(Química-Chang10-ejercicio-6.26) Determine la cantidad de calor (en kJ) que se desprende cuando se producen 1.26 x 104 g de NO2 de acuerdo con la ecuación 2NO(g) + O2(g) → 2NO2 (g) ∆H0 = -114.6 kJ/mol
(Química-Chang10-ejercicio-6.27) Considere la reacción 2H2O(g)→2H2(g)+O2(g) ∆H0 = 483.6 kJ / mol. Si 2.0 moles de H2O (g) se convierten en H2 (g) y O2 (g) contra una presión de 1.0 atm a 125 ° C, ¿cuál es la ∆E para esta reacción?
(Química-Chang10-ejercicio-6.28) Considere la reacción H2(g)+Cl2(g)→2HCl(g) ∆H0 = -184.6 kJ / mol Si 3 moles de H2 reaccionan con 3 moles de Cl2 para formar HCl, calcule el trabajo realizado (en julios) contra una presión de 1.0 atm a 25 ° C. ¿Qué es ∆E para esta reacción? Suponga que la reacción se completa.
(Química-Chang10-ejercicio-6.32) Una pieza de plata de 362 g de masa tiene una capacidad calorífica de 85.7 J / °C. ¿Cuál es el calor específico de la plata?
(Química-Chang10-ejercicio-6.33) Una pieza de metal de cobre de 6.22 kg se calienta de 20.5 °C a 324.3 °C. Calcule el calor absorbido (en kJ) por el metal. Asuma que el calor específico del cobre es 0.39 J/g-°C.
(Química-Chang10-ejercicio-6.34) Calcule la cantidad de calor liberado (en kJ) de 366 g de mercurio cuando se enfría de 77.0 ° C a 12.0 ° C. Asuma que el calor específico del mercurio es 0.14 J/g-°C.
(Química-Chang10-ejercicio-6.37) Una muestra de 0.1375 g de magnesio sólido se quema en un calorímetro de bomba de volumen constante que tiene una capacidad calorífica de 3024 J/°C. La temperatura aumenta en 1.126 ° C. Calcule el calor desprendido por la combustión de Mg, en kJ/g y en kJ/mol.
(Química-Chang10-ejercicio-6.45) ¿Cuál de los siguientes valores de entalpía estándar de formación no es cero a 25 °C? Na(s), Ne(g), CH4(g), S8 (s), Hg(l), H(g)
(Química-Chang10-ejercicio-6.46) Los valores de ∆Hf° de los dos alótropos de oxígeno, O2 y O3, son 0 y 142,2 kJ / mol, respectivamente, a 25 ° C. ¿Cuál es la forma más estable a esta temperatura?
(Química-Chang10-ejercicio-6.47) ¿Cuál es la cantidad más negativa a 25 °C ∆Hf° para H2O (l) o ∆Hf° para H2O (g)?
(Química-Chang10-ejercicio-6.48) Predecir el valor de ∆Hf° (mayor, menor o igual a cero) para estos elementos a 25 °C (a) Br2(g); Br2(l), (b) I2(g); I2(s).
(Química-Chang10-ejercicio-6.49) En general, los compuestos con valores ∆Hf°negativos son más estables que aquellos con valores ∆Hf°positivos. El H2O2(l) tiene un ∆Hf°negativo. Entonces, ¿por qué el H2O2(l) tiene tendencia a descomponerse en H2O(l) y O2(g)?
(Química-Chang10-ejercicio-6.50) Sugiera formas (con ecuaciones apropiadas) que le permitirían medir los valores ∆Hf° de Ag2O(s) y CaCl2(s) a partir de sus elementos. No son necesarios cálculos.
(Química-Chang10-ejercicio-6.51) Calcule el calor de descomposición para este proceso a presión constante y 25 °C: CaCO3(s)→CaO(s)+CO2(g).
(Química-Chang10-ejercicio-6.52a) Las entalpías estándar de formación de iones en soluciones acuosas se obtienen asignando arbitrariamente un valor de cero a los iones H+; es decir, ∆Hf°(H+(aq)) = 0. Para la siguiente reacción HCl(g) → H+(aq)+Cl-(aq) ∆H°=-74.9 kJ/mol, calcule ∆Hf° para los iones Cl-.
(Química-Chang10-ejercicio-6.53a) Calcule el calor de combustión para 2H2(g)+O2(g)→2H2O (l) a partir de las entalpías estándar de formación.
(Química-Chang10-ejercicio-6.53b) Calcule el calor de combustión para 2C2H2(g) + 5O2(g) → 4CO2(g) + 2H2O(l).
(Química-Chang10-ejercicio-6.54a) Calcule el calor de combustión para C2H4(g) + 3O2(g) → 2CO2(g) + 2H2O(l).
(Química-Chang10-ejercicio-6.54b) Calcule el calor de combustión para 2H2S(g) + 3O2(g) → 2H2O(l) + 2SO2(g).
(Química-Chang10-ejercicio-6.56) El cambio de entalpía estándar para la siguiente reacción es 436.4 kJ/mol: H2(g) → H(g) + H(g) Calcule la entalpía estándar de formación del hidrógeno atómico (H).
(Química-Chang10-ejercicio-6.57) A partir de las entalpías estándar de formación, calcule ∆H°rxn para la reacción C6H12(l) → 9O2(g) + 6CO2(g) + 6H2O(l) Para C6H12(l), ∆H°f = -151.9 kJ/mol.
(Química-Chang10-ejercicio-6.59) Determine la cantidad de calor (en kJ) que se desprende cuando se producen 1.26 x 104 g de amoníaco de acuerdo con la ecuación N2(g) + 3H2(g) → 2NH3(g) ∆H°rxn = -92.6 kJ/mol. Suponga que la reacción tiene lugar en condiciones de estado estándar a 25°C.
(Química-Chang10-ejercicio-6.60) A 850 °C, el CaCO3 sufre una descomposición sustancial para producir CaO y CO2. Suponiendo que los valores de ∆H°f del reactivo y los productos son los mismos a 850 °C que a 25 °C, calcule el cambio de entalpía (en kJ) si se producen 66.8 g de CO2 en una reacción.
(Química-Chang10-ejercicio-6.61) A partir de estos datos, S(rómbico) + O2(g) → SO2(g) ∆H°rxn = -296.06 kJ/mol y S(monoclínico) + O2(g) + SO2(g) ∆H°rxn = -296.36 kJ/mol; calcule el cambio de entalpía para la transformación S(rómbica) → S(monoclínica); (Monoclínica y rómbica son formas alotrópicas diferentes de azufre elemental).
(Química-Chang10-ejercicio-6.62) A partir de los siguientes datos, C(grafito) + O2(g) → CO2(g) ∆H°rxn = -393.5 kJ/mol; H2(g) + ½O2(g) → H2O(l) ∆H°rxn = -285.8 kJ /mol; 2C2H6(g) + 7O2(g) → 4CO2(g) + 6H2O(l) ∆H°rxn = -3119.6 kJ/mol; calcule el cambio de entalpía para la reacción 2C(grafito) + 3H2(g) → C2H6(g )
(Química-Chang10-ejercicio-6.64) Calcule el cambio de entalpía estándar para la reacción 2Al(s) + Fe2O3(s) → 2Fe(s) + Al2O3(s) dado que 2Al(s) + 3/2O2(g) → Al2O3(s) ∆H°rxn = -1669.8 kJ/mol; 2Fe(s) + 3/2O2(g) → Fe2O3(s) ∆H°rxn = -822.2 kJ/mol
Química la ciencia central
(Química-Brown13-muestra-5.14b) Una persona de peso promedio usa alrededor de 100 Cal/mi cuando corre o trota. ¿Cuántas porciones de este cereal proporcionan los requisitos de valor de combustible para correr 3 millas? Tenga en cuenta que una porción estándar se cereal proporciona 654 kJ.
LibreChem
(Química-librechem-ejemplo-14.5.1a) En un experimento de calibración, una muestra de ácido benzoico (C6H5COOH) que pesaba 0.825 g se encendió en condiciones idénticas y produjo un aumento de temperatura de 1.94 K. Para el ácido benzoico, se sabe que el calor de combustión a presión constante es 3226 kJ/mol. Utilice este información para determinar la capacidad calorífica del calorímetro ignorando el efecto de los gases.
No hay comentarios:
Publicar un comentario