
Índice
Portada

1. Introducción
|| REGRESAR AL INDICE ||
Un enlace químico es una atracción duradera entre átomos, iones o moléculas que permite la formación de moléculas elementales (como N2), compuestos químicos (como H2O) o iones poliatómicos (NH4+). El enlace puede resultar de la fuerza electrostática entre iones de carga opuesta como en los enlaces iónicos o mediante el intercambio de electrones como en los enlaces covalentes. La fuerza de los enlaces químicos varía considerablemente; hay "enlaces fuertes" o "enlaces primarios", como los enlaces covalentes, iónicos y metálicos, y "enlaces débiles" o "enlaces secundarios", como las interacciones dipolo-dipolo, la fuerza de dispersión de London y los enlaces de hidrógeno.
1.1 Gilbert Newton Lewis
(23 de octubre o 25 de octubre de 1875 - 23 de marzo de 1946) fue un físico químico estadounidense y decano de la Facultad de Química de la Universidad de California. Berkeley. Lewis fue mejor conocido por su descubrimiento del enlace covalente y su concepto de pares de electrones; sus estructuras de puntos de Lewis y otras contribuciones a la teoría del enlace de valencia han dado forma a las teorías modernas del enlace químico. Lewis contribuyó con éxito a la termodinámica química, la fotoquímica y la separación de isótopos, y también es conocido por su concepto de ácidos y bases generalizadas. Lewis también investigó sobre la relatividad y la física cuántica, y en 1926 acuñó el término "fotón" para la unidad más pequeña de energía radiante.
G. N. Lewis nació en 1875 en Weymouth, Massachusetts. Después de recibir su doctorado en química de la Universidad de Harvard y estudiar en el extranjero en Alemania y Filipinas, Lewis se mudó a California en 1912 para enseñar química en la Universidad de California, Berkeley, donde se convirtió en Decano de la Facultad de Química y pasó el resto de su vida. Como profesor, incorporó los principios de la termodinámica en el plan de estudios de química y reformó la termodinámica química de una manera matemáticamente rigurosa accesible a los químicos comunes. Comenzó midiendo los valores de energía libre relacionados con varios procesos químicos, tanto orgánicos como inorgánicos. En 1916, también propuso su teoría de los enlaces y agregó información sobre los electrones en la tabla periódica de los elementos químicos. En 1933, comenzó su investigación sobre la separación de isótopos. Lewis trabajó con hidrógeno y logró purificar una muestra de agua pesada. Luego se le ocurrió su teoría de los ácidos y las bases, y trabajó en fotoquímica durante los últimos años de su vida.

Aunque fue nominado 41 veces, G. N. Lewis nunca ganó el Premio Nobel de Química, lo que resultó en una gran controversia sobre el Premio Nobel. Por otro lado, Lewis fue mentor e influenció a numerosos premios Nobel en Berkeley, incluidos Harold Urey (Premio Nobel de 1934), William F. Giauque (Premio Nobel de 1949), Glenn T. Seaborg (Premio Nobel de 1951), Willard Libby (Premio Nobel de 1960) , Melvin Calvin (Premio Nobel de 1961) y así sucesivamente, convirtiendo a Berkeley en uno de los centros de química más prestigiosos del mundo.
El 23 de marzo de 1946, Lewis fue encontrado muerto en su laboratorio de Berkeley, donde había estado trabajando con cianuro de hidrógeno; muchos postularon que la causa de su muerte fue el suicidio. Después de la muerte de Lewis, sus hijos siguieron la carrera de química de su padre, y el Lewis Hall en el campus de Berkeley lleva su nombre.
(Branch, 1984; Harris, 1999).
2. Generalidades
|| REGRESAR AL INDICE ||
Los átomos de casi todos los elementos se encuentran en combinaciones con otros átomos, ya sea del mismo elemento (ejemplo: O2, N2, Cl2) o de diferentes elementos (ejemplo: H2O, H2SO4). Solo los átomos del grupo 18 “grupo del neón” no se combinan naturalmente (Chang, 2010; Chang & Overby, 2011; Timberlake & Orgill, 2019). Cuando un átomo de un elemento se combina con otro átomo pasa a formar un compuesto, pero cuando lo hace con átomos del mismo tipo forma un elemento, aunque los átomos no se transmutan, cuando estos pasan de estar puros en un elemento para formar un compuesto todas sus propiedades organolépticas cambian, su identidad elemental es alterada para adquirir la identidad del compuesto. Esta propiedad de subsumir las propiedades del elemento y transformarse en algo nuevo y diferente al crear el compuesto fue lo que dio a los antiguos la idea de la transmutación. Sin embargo, las transformaciones químicas son muy limitadas y están gobernadas por leyes físicas muy estrictas, cosa que los alquimistas tardaron casi 2000 años en entender a las malas (Eliade & Ledesma, 1974; Levere, 2001)
Existen dos tipos de compuestos, los compuestos estequiométricos y los compuestos no estequiométricos (Bevan & Hagenmuller, 2013). Los compuestos estequiométricos son el foco de atención de este capítulo, así como las moléculas de elementos y corresponden a sustancias con una composición definida y definible en base a números enteros sencillos (Chang, 2010; Chang & Overby, 2011; Timberlake & Orgill, 2019). Los compuestos no estequiométricos no se ajustan a la regla de los números enteros sencillos (Bevan & Hagenmuller, 2013). Para que dos átomos interactúen entre sí para formar un compuesto o una molécula de elemento, es necesaria la existencia de alguna fuerza que los una, y a esas fuerzas se las denomina enlace químico. Existen varios tipos de enlace, pero lo que es común a todos ellos es la intervención fuerzas dipolares, en otras palabras, el modelo de trabajo mental serán los IMANES, si usted sabe cómo funciona un imán, sabrá cómo funciona la química.

Figura 2.1. Entender el fenómeno de atracción/repulsión es vital para toda la química.
2.1 Definición general del enlace químico
Un enlace químico es una atracción permanente de dos núcleos atómicos que permite la formación de combinaciones químicas conocidas como compuestos (Chang, 2010; Chang & Overby, 2011; Timberlake & Orgill, 2019). La atracción tiene un punto límite, es decir, los dos átomos se moverán juntos atraídos, pero con una distancia mínima en la cual empezaran a repelerse mutuamente, esta fuerza que impide que las moléculas colapsen y los núcleos se fusionen se denomina fuerza nuclear (Bader, Hernández‐Trujillo, & Cortés‐Guzmán, 2007). Esta ambivalencia entre la atracción que forma la molécula del compuesto y la repulsión que evita que la materia colapse depende de las partículas subatómicas del átomo y del viejo juego de las cargas.
Las cargas similares se repelerán
Las cargas opuestas se atraerán
Siempre les digo a mis estudiantes que la química es falsamente compleja, ya que en el fondo muchos de los problemas se resuelven fácilmente si se conoce las reglas de atracción electrostáticas, las cuales son las mismas que cuando se tiene un par de imanes. Todos los enlaces químicos dependen del juego de las cargas, alternando polos o posiciones positivas con posiciones negativas. Un átomo normal posee tres partículas básicas, los protones, los neutrones y los electrones. Los protones se encuentran en el núcleo y generan una carga electrostática positiva, por lo que atraen la carga opuesta que es emitida por los electrones. Los neutrones no tienen carga y por lo tanto no nos interesan tanto en (Chang, 2010; Chang & Overby, 2011; Timberlake & Orgill, 2019).
2.2 Tipos de enlaces
Esta alternancia de positivo y negativo puede permitir que un electrón sirva como puente de forma tal que un átomo (+) esté atraído por un electrón (-) que a su vez es atraído por un átomo (+), esta alternancia de cargas puede repetirse en estructuras más complejas, pero es en esencia la misma. Aunque los electrones son la base de cualquier enlace químico, ya que son los portadores de la carga negativa, el modo en que enlazan a los átomos varía en intensidad y potencia, por lo que hemos tenido que clasificar los enlaces químicos como fuertes y débiles, y cada una de estas dos a su vez posee otra serie de subcategorías, aunque en el fondo de todas ellas siempre se encontrará el juego de las cargas.

Figura 2.2. Los tres tipos de enlace comunes en muchas sustancias, estos dependen de la electronegatividad de los núcleos involucrados, a mayores diferencias entre la electronegatividad de los núcleos, más iónico será el enlace.
En general, los enlaces químicos fuertes están asociados a compartir electrones entre núcleos o a parejas de cargas electrostáticas muy potentes “polos iónicos” que mantienen los núcleos fijos en su posición molecular en gran medida. Los átomos en moléculas, cristales, metales y gases diatómicos se encuentran unidos por enlaces fuertes. Por otro lado, los enlaces débiles, al igual que en los anteriores son causados por cargas electrostáticas generadas por electrones, pero en este caso la presencia del electrón genera un polo negativo débil que atrae levemente los núcleos de otros átomos. Debido a la debilidad los núcleos solo se atraen ligeramente, como si fuera una tendencia estadística, pero no se mueven juntos “a menos que estén en estado sólido”, en otras palabras, no se forman moléculas.
Aunque todos los tipos de enlace son matematizables por la mecánica cuántica, en la práctica aprenderemos una serie de reglas simples que permiten predecir muchas moléculas, aunque no todas. Las predicciones tienen diferentes grados de especificidad y complejidad, y van desde la cantidad de átomos en una molécula hasta la organización tridimensional de esta (Chang, 2010; Chang & Overby, 2011; Timberlake & Orgill, 2019).
3. Historia
|| REGRESAR AL INDICE ||
3.1 Los griegos
Demócrito y Leucipo argumentaban que los átomos de las sustancias sólidas se encontraban enganchados, lo cual les permitía moverse juntos creando la ilusión de un objeto único. Una característica de dicho enganche es que debería mantener las distancias relativas de cada átomo constante de forma que la estructura enganchada mantuviera su forma al entrar en contacto con otros átomos (Bantz, 1980).
3.2 Lucrecio
Lucrecio extendió los razonamientos de Demócrito hacia el estado líquido, asumiendo que los átomos poseían una especie de superficie, de esta forma los líquidos deberían estar compuestos por átomos lisos que jamás se enganchan entre sí, mientras que los átomos de los sólidos tenían estructuras de superficie que les permitía acoplar de forma específica (Dunitz & Gavezzotti, 2009; Mackay, 2002).
Este razonamiento permitirá explicar cómo líquidos con alta cohesión como el aceite existían, asumiendo que esos átomos eran una especie de intermedio, siendo lisos, pero con algunos enganches que hacían que el líquido tuviera una cohesión más alta que otros como el agua o el alcohol (Dunitz & Gavezzotti, 2009; Mackay, 2002). Sin embargo, debido a la autoridad de Aristóteles, la teoría atómica sería suprimida durante unos cuantos siglos, y en consecuencia también las especulaciones sobre en enlace químico (Dunitz & Gavezzotti, 2009; Mackay, 2002).
3.3 El siglo XVIII
Aunque estamos acostumbrados a pensar que Dalton fue quien revivió la teoría atómica, ya en el siglo XVIII tenemos unas especulaciones bastante serias de la existencia del átomo y del enlace entre átomos de la mano de filósofos naturales tan influyentes como Isaac Newton (Pyle, 1995), aunque otros autores también razonaron al respecto como: William Cullen, Roger Boscovich, Torbern Bergman, William Higgins o Robert Boyle (Lindsay, 2009). Esta versión del átomo extraía directamente de las ideas de Lucrecio y Democrito imaginaba a los átomos con diferentes formas en sus superficies, lo cual les permitiría engancharse entre sí. El problema radicaba en que las sustancias se unen entre sí de formas complejas, tanto así que aun las actuales teorías del átomo tienen problemas para predecir todos los enlaces posibles. La importancia o problema que trajo esto fue que los alquimistas o filósofos naturales que intentaron sistematizar las combinaciones de sustancias por medio de una teoría de superficies de átomos fracasaron de forma estrepitosa (Kuhn, 1945).
3.4 El siglo XIX
En 1819 gracias al invento de la pila voltaica Berzelius desarrolló una teoría del enlace que se basaba en ideas del electromagnetismo, en otras palabras, de la atracción electrostática de dos polos opuestos. Para la mitad del siglo XIX una serie de científicos encabezados por Edward Frankland, pero dentro de los cuales también se puede destacar a Kekulé, Couper, Butlerov y Kolvbe desarrollaron en conjunto el gran concepto de Valencia, aunque en aquel entonces se lo conocía por el más descriptivo término “Poder de Combinación” (Larder, 1971).
En lo personal preferiría que se hubiera mantenido el término “poder de combinación” en lugar de valencia ya que te indica de forma más intuitiva para que sirve esa propiedad periódica. El poder de combinación implicaría la cantidad de sustancia que podía combinarse con otras cantidades estandarizadas que denominamos moles. En un lenguaje más atomista la valencia indica la capacidad de combinación de un átomo con otros átomos.
3.5 Siglo XX pre cuantico
Uno de los conceptos más alabados en la química del enlace es la regla del octeto, conocida originalmente como la ley de Abegg (Jensen, 1984). La ley de Abegg fue propuesta en 1904 por el químico alemán Richard Abegg, la cual establecía que la diferencia máxima positiva o negativa de valencia de un elemento es frecuentemente de ocho. En palabras más modernas se interpreta en el sentido de que los elementos ganan o pierden electrones de forma tal que su última capa de electrones (s, p) completa ocho electrones, de forma tal que se asemejan a su gas noble más cercano. En 1916 el químico Gilbert N. Lewis desarrolló el concepto de par de electrones enlazantes, en la cual un átomo puede compartir electrones con otro átomo para poder cumplir la regla del octeto. Un átomo puede compartir con otro átomo hasta seis electrones formando enlaces simples, dobles o triples. En otras palabras, Lewis expandió la idea de los octetos (Jensen, 1984).
Las representaciones originales no son para nada parecidas a las que empleamos en la actualidad, ya que originalmente Lewis trabajó con átomos en forma de cubo en su capa externa, los electrones se encontraban fijos en las aristas del cubo, lugar donde “se enganchaban” los átomos para formar moléculas compuestas por varios cubos (Kohler, 1971; Niaz, 2001). En 1916 Walter Kossel publicó una teoría semejante a la de Lewis, solo que en su modelo los electrones no se compartían formando puentes de unión, sino que eran transferidos completamente, de forma tal que la unión se generaba por la atracción electrostática de un átomo que se tornaba negativo con otro que se tornaba positivo (Christe, 2013; Soukup, 2005). De esta forma obtenemos los conceptos básicos que enseñamos en el tema del enlace químico, los enlaces covalente “Lewis” y iónico “Kossel” ambos organizados de forma tal que respectan la ley de Abegg “octeto”.
3.6 La teoría cuántica
La versión del enlace químico que enseñamos es una extraña mezcla entre los conceptos pre-cuánticos y los conceptos cuánticos, esto se debe probablemente a que el enlace químico a la vista del modelo cuántico es un poco más difícil de interpretar, después de todo hay más matemáticas y de las complejas (Bakowies & Thiel, 1996; Cremer & Kraka, 1984). En cualquier caso, para 1927 el físico danés Oyvind Burrau describió por primera vez el enlace covalente perfecto de la molécula de hidrógeno gaseoso. Este trabajo demostró que una aproximación desde la teoría mecánico-cuántica del átomo era posible con resultados cuantitativamente precisos, aunque el problema seguía siendo el poder describir moléculas con más de un electrón (Rhodes & Macrae, 2015).
Debido a la complejidad matemática del asunto se emplearon aproximaciones menos rigurosas en el mismo año por parte de Walter Weitler y Fritz London. El método de Heitler-London conformaría la base de una de las visiones predominantes del enlace químico moderno, es decir la Teoría del Enlace de Valencia (Hund, 1977). En 1929 se propuso las aproximaciones linear de la combinación de orbitales atómicas y moleculares por parte de Sir John Lennard-Jones, la cual será conocida en la actualidad simplemente como la teoría del orbital molecular. En la actualidad se emplean las dos teorías del enlace lo cual demuestra que debe existir alguna otra explicación más general, pero que no ha sido formulada aun (Lennard-Jones, 1949).
Este breve resumen de la historia del enlace químico nos permite hacer una serie de afirmaciones. Los modelos del enlace químico son todos imperfectos, y cualquiera que enseñemos tendrá una serie de problemas, moléculas no explicadas y errores garrafales si se toman los ejemplos donde no aplican. El punto importante es dar una visión del enlace químico que nos permita entender el modo en que se combinan los átomos, en otras palabras, poder describir el poder de combinación de los átomos y que propiedades poseen en términos cualitativos, sin tener que meternos demasiado en la mecánica cuántica. En otras palabras, hay que tratar de generar un modelo funcional.
4. Enlace fuerte
|| REGRESAR AL INDICE ||
Los enlaces fuertes son fuerzas intermoleculares que mantienen unidos los átomos en las moléculas. Los enlaces fuertes se forman ya sea por compartir electrones generando una unión o mediante la transferencia de electrones que genera una atracción electrostática. Existen varios tipos generales de enlaces fuertes, aunque en realidad deberíamos hablar de dos: Iónico-Covalente y Metálico (Chang & Overby, 2011; Chang, 2006; Ebbing & Gammon, 2008; McMurry, Castellion, & Ballantine, 2007; Timberlake, 2015).
4.1 Enlace iónico
Regla heurística rápida: Enlace formado entre un metal y un no metal. El enlace iónico es un tipo de enlace químico que involucra la atracción electrostática entre dos iones opuestos que funcionan como los dos polos de un dipolo magnético “un imán”. La naturaleza del enlace iónico es la que le da sus propiedades a los compuestos iónicos y a los iones. En el enlace iónico uno de los átomos cede un electrón tornándose positivo, mientras que otro átomo adquiere un electrón, tornándose negativo. Cuando los átomos han adquirido o perdido electrones se los denomina iones. Los positivos reciben el nombre de CATIONES y los negativos de ANIONES. La transferencia casi total de los electrones se denomina electrovalencia, ya que el poder de combinación es mediado por la carga electrostática y no por compartir electrones (Chang & Overby, 2011; Chang, 2006; Ebbing & Gammon, 2008; McMurry et al., 2007; Timberlake, 2015).

Figura 4.1. El empaquetamiento de las sustancias iónicas generan cristales muy estables, por lo que tienden a ser sólidos duros y cristalinos.
Por norma general los metales tienen la tendencia a perder electrones, mientras que los no metales tienden a atraerlos, sin embargo, esas tendencias dependen de propiedades diferentes del poder de combinación de los elementos, de allí que la mera regla del octeto no nos ayuda a predecir muchas situaciones. Los iones pueden son monoatómicos o poliatómicos, lo que importa es la naturaleza monopolar del ion debido a haber adquirido o perdido un electrón (Chang & Overby, 2011; Chang, 2006; Ebbing & Gammon, 2008; McMurry et al., 2007; Timberlake, 2015).
👉 El continuo iónico – covalente: extremo iónico
El continuo iónico covalente es una idea que emerge de la naturaleza del enlace químico fuerte, y es que no hay un núcleo atómico lo bastante fuerte como para despojar de la nube de electrones a otro átomo de forma permanente. Un núcleo muy fuerte como el Flúor puede transferir el electrón de otro átomo hacia sí mismo la mayoría del tiempo, pero en algún microinstante el electrón puede transferirse al núcleo del elemento más débil, por lo que no existe un enlace iónico perfecto.
En otras palabras, todo enlace iónico posee algún nivel de covalencia, es decir de compartir electrones entre los núcleos, estamos hablando por lo tanto de una propiedad cualitativa con una escala continua y no de una propiedad cualitativa de blanco o negro. Ahora la pregunta es ¿Cómo diferenciar un enlace iónico de un enlace covalente? Existen dos formas de responder esta pregunta la mayoría de las veces, pero el lector deberá tener en cuenta que algunas moléculas pueden no comportarse de acuerdo con los siguientes lineamientos:
👉 Cuando el valor absoluto de la diferencia de electronegatividades |Δχ| es superior a 1.7.
👉 Dado que los metales tienen bajas electronegatividades y los no metales altas electronegatividades lo anterior se puede resumir con que, se forman enlaces iónicos entre metales y no metales, siendo los metales quienes asumen la carga positiva y los no metales la carga negativa.
👉 Propiedades del enlace iónico
El enlace iónico es fuerte pero inflexible, es muy frágil, de forma tal que en presencia de agua o cualquier solvente polar tienen la tendencia a romperse. Una vez rotos y en solución, los compuestos iónicos son capaces de conducir la electricidad con facilidad, pero el sólido no lo hace. Los puntos de fusión tienden a depender de la diferencia de electronegatividades, a mayor diferencia más fuerte el enlace, más difícil romperlo con calor y por lo tanto funden a mayor temperatura, o se solubilizan más difícilmente. Sin embargo, hay que tener en cuenta que muchas de esas tendencias tienen excepciones notables. La geometría molecular del enlace iónico sigue las reglas del máximo empaquetamiento posible (Imagen anterior) debido a que no existen pares de electrones que se repelan mutuamente. Sin esos “espacios ocupados” los átomos iónicos pueden empaquetarse de la forma más eficiente posible.
4.2 Enlace covalente
Regla heurística rápida: enlace formado entre no metales. Un enlace covalente es un tipo de enlace que involucra la formación de una pareja de electrones compartida por dos núcleos atómicos, en este caso ninguno de los dos átomos adquiere una naturaleza polar fuerte, por lo que la atracción se da por la atracción electrostática del par de electrones enlazantes. A diferencia del enlace iónico, el encale covalente tiene diferentes subtipos como el enlace pi, el enlace sigma, enlace deslocalizado o el enlace covalente polar. El termino covalente se origina en 1932 y señala un tipo de enlace de valencia compartida.
👉 El continuo iónico – covalente: extremo covalente
El continuo iónico covalente no tiene un extremo iónico perfecto, pero si tiene un extremo covalente perfecto. Dos átomos con la misma electronegatividad pueden unirse entre sí al compartir de forma perfecta dos electrones, sin que uno lo tenga más tiempo que el otro. A partir de aquí uno puede empezar a decir que cualquier enlace donde exista una diferencia de electronegatividades, así sea leve tendrá algunas características del enlace iónico, y de hecho así es, por tal razón en el continuo iónico covalente emerge un tercer concepto intermedio llamado covalente polar. El enlace covalente polar se caracteriza porque, aunque los electrones se comparten, ellos tienden a pasar más tiempo alrededor del núcleo más electronegativo creando un dipolo leve, con una carga parcial negativa en el átomo más electronegativo y una carga parcial positiva en el extremo menos electronegativo, lo cual hace que estas sustancias no se comporten idealmente en pequeños instantes.
👉 Enlace covalente perfecto
Regla heurística rápida: enlace formado entre átomos de un mismo elemento. También llamado NO polar, es aquel formado por núcleos con la misma electronegatividad, estas moléculas no forman dipolos por lo que no pueden interactuar con otras moléculas con facilidad, por lo que no son solubles y tienden a ser gases. ¿Cómo identificar un enlace covalente perfecto? Existen dos formas de responder esta pregunta la mayoría de las veces, pero el lector deberá tener en cuenta que algunas moléculas pueden no comportarse de acuerdo a los siguientes lineamientos:
👉 Cuando el valor absoluto de la diferencia de electronegatividades |Δχ| es superior a 0. Para que la igualdad anterior se cumpla, lo común es que el enlace se forme entre átomos de un mismo elemento, como entre dos hidrógenos, dos oxígenos o incluso entre más de dos átomos de un mismo elemento.
👉 Dado lo anterior, siempre que observemos una molécula formada por dos o más átomos del mismo elemento como: H2, O2, Cl2, S8, C60; definiremos que su enlace es covalente.
👉 También serán covalentes los enlaces entre carbonos de una molécula orgánica.
👉 Enlace covalente polar
La diferencia de electronegatividades forma un dipolo magnético débil, por lo que la molécula funciona como un imán débil, en esencia tiene lo mejor de los dos extremos, posee los polos de una molécula iónica, pero no se rompe con facilidad, por lo que son moléculas estables. ¿Cómo identificar un enlace covalente polar? Existen dos formas de responder esta pregunta la mayoría de las veces, pero el lector deberá tener en cuenta que algunas moléculas pueden no comportarse de acuerdo a los siguientes lineamientos:
👉 Cuando el valor absoluto de la diferencia de electronegatividades |Δχ| es inferior a 1,7.
👉 Lo anterior se cumple comúnmente cuando los átomos involucrados en el enlace son ambos no-metales, pero de elementos diferentes.
👉 Simples y múltiples
Tanto los enlaces covalentes perfectos como los enlaces covalentes polares pueden presentarse como únicos entre dos átomos o múltiples, siendo generalmente un máximo de tres enlaces entre dos núcleos. Al primer enlace entre dos núcleos se lo denomina sigma y puede girar sobre sí mismo, mientras que los enlaces restantes se les denomina pi y son rígidos.
👉 Enlace resonante o deslocalizado
Este se caracteriza por una molécula con un electrón sobrante como en los aniones o cationes poliatómicos. Este electrón puede formar un enlace doble o triple en cualquiera de las posiciones donde ya hay un enlace, lo cual genera una serie de posibles fórmulas, todas válidas y que probablemente se manifiestan al mismo tiempo resonando entre unas y otras, aunque posiblemente tampoco exista ninguna realmente.

Figura 4.2. La resonancia implica generalmente enlaces dobles y simples que se alternan al azar, generando varias estructuras plausibles.
La visión alternativa sobre estos electrones sobrantes o faltantes es que hay una deslocalización de la posición del electrón, y de esta forma generando una estructura molecular que no podemos representar fácilmente. Dos ejemplos de enlace con electrón deslocalizado son el enlace aromático y el enlace metálico.
👉 Enlace metálico
Bien, ese es el primer enlace fuerte en el que se presentan dos conceptos clave, primero, es un enlace en el que no intervienen dolo 2 núcleos y un par de electrones por enlace, en su lugar tienes miles de millones de núcleos, con miles de millones de electrones moviéndose de manera laxa entre toda la masa, generando una nube, a este tipo de movimiento lo llamamos deslocalización. La deslocalización del enlace metálico hace que los electrones se muevan fácilmente entre la gran nube de probabilidades, lo que explica la gran conductividad eléctrica de los metales, pues debido a la deslocalización de sus electrones, estos se mueven muy fácilmente o pueden ser empujados fácilmente.
Adicionalmente los núcleos y electrones pueden vibrar con facilidad, y cuando una masa de átomos vibra manifiesta calor, eso implica que el enlace metálico puede distribuir o conducir calor con facilidad. La representación de un metal puro es en consecuencia la de una sustancia cuyo estado de oxidación es neutro, es decir cero.
5. Enlace débil
|| REGRESAR AL INDICE ||
También conocidas como fuerzas intermoleculares o interacciones intermoleculares, son una serie de fuerzas de atracción o de repulsión que actúan entre las partículas, sean estos átomos, moléculas o iones. Estas interacciones son débiles cuando se las compara con las fuerzas intermoleculares también conocidas como los enlaces químicos fuertes. Sin embargo, pueden afectar de forma significativa las propiedades físicas de una sustancia una vez que se suman de forma neta. Por ejemplo, en las moléculas orgánicas a medida que una cadena de carbonos se hace más larga, también cambia el estado de la materia en la cual se presenta, de esta forma las cadenas muy cortas son gases, las medianas son líquidos y las muy largas son ceras o sólidos.
La investigación sobre las interacciones moleculares inició poco después de haberse propuesta la ley de los gases ideales, y era responder una pregunta simple, ¿Por qué los gases no se comportan idealmente? Uno de los presupuestos de la ley es que las partículas al impactar no se atraen o repelen, no interactúan entre sí, y el hecho de que los gases reales no se comporten idealmente implicaba necesariamente la idea de que al impactar algunas partículas se atraen y otras se repelen. Ya en el siglo XIX varios científicos de prestigio hicieron grandes avances en el entendimiento de dichas fuerzas como: Clairaut, Laplace, Gauss, Maxwell y Boltzmann. Las fuerzas intermoleculares atractivas se distinguen en dos categorías principales: las interacciones dipolo-dipolo; y fuerzas de van der Waals.

Figura 5.1. Los puentes de hidrógeno son interacciones típicas entre dos moléculas con dos polos, en ese sentido los polos opuestos se atraen.
5.1 Interacciones dipolo-dipolo
Las interacciones dipolo-dipolo son atracciones electrostáticas entre dipolos permanentes en las moléculas. Debido a las geometrías moleculares las moléculas covalentes polares pueden experimentar un comportamiento semejante a un imán débil, y todo imán tiene dos polos, uno negativo y otro positivo, de allí surge el nombre dipolo. En ese orden de ideas las interacciones entre dipolos no son diferentes a las interacciones que experimentarían dos imanes, y en consecuencia los polos opuestos se atraerán y los polos iguales se repelerán.
5.2 Iones-dipolo
Al interior de las interacciones dipolo-dipolo se encuentran las interacciones ion-dipolo las cuales no son muy diferentes en naturaleza, la única diferencia es que el ion se comporta como un monoimán o mejor dicho como un monopolo, pero debido a que su carga electrostática en ese polo es más fuerte atraerá con más potencia los polos opuestos de los dipolos que lo rodean.

Figura 5.2. Los iones con carga permanente negativa pueden atraer los hidrógenos del agua, lo cual les permite diluirse con facilidad.
5.3 Puentes de hidrógeno
Los puentes de hidrogeno o enlaces de hidrógeno son una atracción de tipo dipolo-dipolo/ion-dipolo entre el hidrogeno unido a una molécula/ion contra el polo negativo de otra molécula o ión (los ejemplos anteriores también son puentes de hidrógeno ya que el extremo positivo del enlace débil es un hidrógeno). Típicamente el hidrogeno va a ser atraído contra elementos muy electronegativos que además hibridan dejando electrones no enlazantes que sin embargo generan una carga electrostática negativa como el nitrógeno, el oxígeno o el flúor. El puente de hidrogeno es comúnmente descrito como una atracción electrostática fuerte, tanto así que puede llegar a presentar características de enlace covalente al ser direccional, ser más fuerte de las interacciones de van der Walls e involucrar pocos puntos de unión. De hecho, el amoniaco posee un punto electrostático tan fuerte en forma de sus dos electrones no enlazantes que son capaces de unir un protón libre en el agua formando el ion amonio en lo que se conoce como enlace covalente coordinado.
5.4 Solubilidad de los dipolos y monopolos
Una de las propiedades físicas que más se afectan por la existencia de las interacciones de dipolos o monopolos es la solubilidad. Las sustancias polares son aquellas que exhiben algún tipo de polaridad ya sea dipolar o monopolar como en los iones. El agua al tener mucho hidrógeno puede enlazar por puente de hidrogeno a cualquier molécula que exhiba interacciones polares integrándolas a una red de atracciones. Por el contrario, las moléculas que no pueden enlazarse por puente de hidrogeno son excluidas de la red de atracciones siendo en consecuencia insolubles en el agua.
5.5 Estado de la materia de los dipolos
Un dipolo es más difícil de separar de otros dipolos, eso implica que una molécula dipolar va a agruparse con sus compañeras, por lo que hay que calentarlas más para que ingresen a la fase líquida o sólida. Este detalle explica porque el agua es líquida y no un gas a la temperatura ambiente del planeta Tierra. Otras moléculas como el metano no son polares, no se unen entre si y por lo tanto no se unen espontáneamente.
5.6 Los dipolos y la vida
Las moléculas responsables por nuestra existencia, los ácidos nucleicos y las proteínas son capaces de entablar interacciones moleculares por puente de hidrogeno, las cuales fuerzan a la cadena a tomar formas complejas que les otorgan sus funciones vitales. En las proteínas por ejemplo, las interacciones dipolo-dipolo son responsables por la estructura secundaria, terciaria y cuaternaria.
5.7 Interacciones de van der Waals
Las interacciones de van der Waals también son electrostáticas, pero más débiles. La característica principal de la interacción de van der Waals es que una de las moléculas es apolar o no polar, pero aun así puede ser atraída o repelida por interacciones electrostáticas que se supone no debería tener, y por lo tanto son más débiles.

Figura 5.3. Existen tres maneras de alterar a una molécula covalente, la primera es que un ion permanente lo induzca; la segunda es que una molécula con un polo negativo lo induzca; y la tercera a inestabilidades al azar debido a la indeterminación cuántica propia de los electrones.
Algunas como la interacción de Debye se deben a que un dipolo permanente desestabiliza a una molécula apolar creando un dipolo inducido momentáneo que atrae o repele. En otros casos un ion puede desestabilizar a la molécula apolar fenómeno denominado como ión contra dipolo inducido. Sin embargo, las fuerzas dominantes son las interacciones de London, las cuales juegan con el principio de indeterminación de la quántica. Debido a la naturaleza aleatoria del movimiento de los electrones, existen leves momentos en los que la distribución electrónica en un enlace covalente perfecto no es perfecta, creando un breve momento dipolar y por lo tanto un leve momento de atracción-repulsión.
Aunque este momento de atracción es despreciable en moléculas pequeñas, a medida que las moléculas se hacen más largas la atracción de London se hace más importante. Esta es la interacción que hace que las moléculas orgánicas largas tiendan a ser sólidos y las moléculas cortas sean gases, ya que a mayor longitud la probabilidad de que en algún punto se genere un dipolo aleatorio momentáneo se incrementa.
6. Símbolos de Lewis y el octeto
|| REGRESAR AL INDICE ||
Los electrones involucrados en el enlace químico son los electrones de valencia, que, para la mayoría de los átomos, son los que se encuentran en la capa más externa ocupada. El químico estadounidense G. N. Lewis (1875-1946) sugirió una manera simple de mostrar los electrones de valencia en un átomo y rastrearlos durante la formación de enlaces, utilizando lo que ahora se conoce como símbolos de puntos de electrones de Lewis o simplemente símbolos de Lewis. El símbolo de Lewis para un elemento consta del símbolo químico del elemento más un punto para cada electrón de valencia. El azufre, por ejemplo, tiene la configuración electrónica Ne3s23p4 y, por lo tanto, seis electrones de valencia.

Figura 6.1. Modelo de puntos de Lewis para el átomo de azufre
Los puntos se colocan en los cuatro lados del símbolo (arriba, abajo, izquierda y derecha) y cada lado puede acomodar hasta dos electrones. Los cuatro lados son equivalentes, lo que significa que la elección de en qué lado colocar dos electrones en lugar de uno es arbitraria. En general, separamos los puntos tanto como sea posible. En el símbolo de Lewis para S, por ejemplo, preferimos la disposición de puntos que se muestra en lugar de la disposición que tiene dos electrones en tres de los lados y ninguno en el cuarto. Las configuraciones electrónicas y los símbolos de Lewis para los elementos del grupo principal de los períodos 2 y 3 se muestran en la tabla:
Tabla 6‑1. Configuraciones electrónicas y modelos de punto de Lewis para los elementos de los periodos 2 y 3 de la tabla periódica.

Observe que el número de electrones de valencia en cualquier elemento representativo es el mismo que el número de grupo del elemento empleando la nomenclatura CAS, aunque no concordará con las nomenclaturas de grupo de la IUPAC. Por ejemplo, los símbolos de Lewis para oxígeno y azufre, miembros del grupo 6ª en la nomenclatura CAS, muestran seis puntos.
6.1 La regla del octeto
Ten en cuenta que se denomina regla y no ley por una razón fundamental, tiene una enorme cantidad de excepciones, sin embargo, hace parte importante de la historia de la química que ha sido traducida a los libros de texto y por ende es un concepto que es irrenunciable, aunque en lo personal no me gusta.
Los átomos a menudo ganan, pierden o comparten electrones para lograr la misma cantidad de electrones que el gas noble más cercano a ellos en la tabla periódica. Los gases nobles tienen arreglos de electrones muy estables, como lo demuestran sus altas energías de ionización, baja afinidad por electrones adicionales y falta general (pero no total como lo revelan algunos compuestos de xenón) de reactividad química. Debido a que todos los gases nobles, excepto el He, tienen ocho electrones de valencia, muchos átomos que experimentan reacciones terminan con ocho electrones de valencia. Esta observación ha llevado a una directriz conocida como la regla del octeto: los átomos tienden a ganar, perder o compartir electrones hasta que están rodeados por ocho electrones de valencia.
Una primera excepción son los elementos cuyo gas noble más cercanos el helio, allí la regla del octeto pasa a ser de dueto, es decir los elementos tendrán la tendencia a configurarse en dos electrones de valencia en su capa más externa.
Un octeto de electrones consta de subcapas s y p completas en un átomo. En un símbolo de Lewis, un octeto se muestra como cuatro pares de electrones de valencia dispuestos alrededor del símbolo del elemento, como en los símbolos de Lewis para Ne y Ar. Hay excepciones a la regla del octeto, como veremos más adelante en el capítulo, pero proporciona un marco útil para introducir muchos conceptos importantes de enlace.
6.2 Estructuras de Leweis para compuestos iónicos
Las sustancias iónicas generalmente resultan de la interacción de los metales en el lado izquierdo de la tabla periódica con los no metales en el lado derecho (excluyendo los gases nobles). Por ejemplo, cuando el sodio metálico, Na(s), si se pone en contacto con cloro gaseoso, Cl2(g), y un agente desestabilizante de la estructura iónica del metal, como una gota de agua se produce una reacción violenta. El producto de esta reacción muy exotérmica es el cloruro de sodio, NaCl(s): Na(s) + Cl2(g) → NaCl(s) ∆Hf° = —410.9 kJ/mol.
El cloruro de sodio se compone de iones Na+ y Cl- dispuestos en una matriz tridimensional. La formación de Na+ a partir de Na y Cl- a partir de Cl2 indica que un átomo de sodio ha perdido un electrón y un átomo de cloro lo ha ganado; decimos que ha habido una transferencia de electrones del átomo de Na al átomo de Cl.
Tres de las propiedades atómicas nos dan una indicación de la facilidad con la que ocurre la transferencia de electrones: la energía de ionización, que indica la facilidad con la que se puede eliminar un electrón de un átomo; la afinidad electrónica, que mide cuánto quiere un átomo ganar un electrón; y la electronegatividad que indica la facilidad con la que un núcleo atómico atrae electrones propios o ajenos.
La transferencia de electrones para formar iones de carga opuesta ocurre cuando un átomo cede fácilmente un electrón (baja energía de ionización/ baja electronegatividad) y otro átomo gana fácilmente un electrón (alta afinidad electrónica / alta electronegatividad). Así, el NaCl es un compuesto iónico típico porque consiste en un metal de baja energía de ionización y un no metal de alta afinidad electrónica. Usando símbolos de puntos de electrones de Lewis (y mostrando un átomo de cloro en lugar de la molécula de Cl2), podemos representar esta reacción como:

Figura 6.2. Modelo de puntos de Lewis para la ionización de sodio metálico y cloro elemental.
La flecha púrpura indica la transferencia de un electrón del átomo de Na al átomo de Cl. Cada ion tiene un octeto de electrones, siendo el octeto de Na+ los electrones 2s22p6 que se encuentran debajo del único electrón de valencia 3s del átomo de Na. Hemos puesto un paréntesis alrededor de ambos iones para enfatizar que han dejado de ser átomos libres y se han transformado en iones con cargas electrostáticas permanentes.
Las sustancias iónicas poseen varias propiedades características. Suelen ser sustancias quebradizas con puntos de fusión elevados. Suelen ser cristalinos. Además, los cristales iónicos a menudo se pueden escindir; es decir, se separan a lo largo de superficies lisas y planas. Estas características son el resultado de fuerzas electrostáticas que mantienen los iones en una disposición tridimensional rígida y bien definida.
6.3 Formación del enlace iónico
Aunque no siempre sucede, dependiendo de particularidades de cada elemento, lo común es que si tenemos iones positivos e iones negativos, esos actúen como enormes monopolios qué se atraen mutuamente hice auto organizan en una estructura estable cristalina que intercala la posición positiva y la posición negativa de manera tridimensional, en otras palabras se genera el enlace iónico.

Figura 6.3. Modelo que representa el enlace iónico.
Observe que cuando representamos el enlace iónico no dibujamos un enlace entre los 2 átomos pues no existe, lo que une a los átomos es la carga electrostática opuesta, no el hecho de que existan electrones en medio de ellos que los mantengan unidos.
6.4 Configuración electrónica de un ion en elementos de grupos representativos
La energía de la formación de enlaces iónicos ayuda a explicar por qué muchos iones tienden a tener configuraciones electrónicas de gas noble. Por ejemplo, el sodio pierde fácilmente un electrón para formar Na+, que tiene la misma configuración electrónica que el Ne:

Figura 6.4. cambio en la configuración electrónica del sodio cuando éste se ioniza.
Aunque la energía reticular aumenta con el aumento de la carga iónica, nunca encontramos compuestos iónicos que contengan iones Na2+. El segundo electrón eliminado tendría que provenir de una capa interna del átomo de sodio, y eliminar electrones de una capa interna requiere una gran cantidad de energía. El aumento de la energía de la red no es suficiente para compensar la energía necesaria para eliminar un electrón de la capa interna. Por lo tanto, el sodio y los otros metales del grupo 1A se encuentran en sustancias iónicas solo como iones (1+).
Aunque en el libro de textos nos dice que para ello un cloro el caso similar y nos pone en un caso hipotético para los iones cloruro(1-) y el hipotético Cl(2-), con un argumento energético, el cual es correcto, omite un gran detalle, el cloro a diferencia del sodio puede hibridarse, que es la capacidad que tienen los orbitales de combinarse con otros para alterar su capacidad de perder o capturar electrones.
Si el cloro siguiera la ley del octeto a rajatabla sólo debería tener un estado de oxidación posible (-1), y lo único que podría hacer es capturar da un electrón por cloro, sin embargo, el cloro también es capaz de perder electrones, y esa capacidad para perder electrones no es explicada por la ley del octeto.
Sin embargo, en términos de resolución de ejercicios de lápiz y papel lo que nos van a preguntar es cuál es el ion que se va a generar con mayor probabilidad, siendo estos iones monoatómicos, y en tales casos la regla del octeto sí que es útil. En consecuencia para ejercicios de lápiz y papel vamos a aplicar el siguiente algoritmo de solución:
👉 el ion más probable va a ser aquel que es semejante a su gas noble más cercano.
👉 para metales de los grupos representativos (ignorando metales de transición y tierras raras) eso implica retirar los electrones que quedan sobrando desde la configuración electrónica del gas noble anterior, luego agregar la magnitud de esos electrones como cargas positivas en súper índices con una estructura en la cual se coloca primero la magnitud y luego el símbolo positivo, ignorando los uno.
👉 para no metales eso implica agregar los electrones que quedan faltando para alcanzar a su gas noble siguiente, luego agregar la magnitud de esos electrones como cargas negativas en súper índices con una estructura en la cual se coloca primero la magnitud y luego el símbolo negativo, ignorando los uno.
Química la ciencia central 13
Muestra 8.2. Prediga el ion generalmente formado por (a) Sr, (b) S, (c) Al
Muestra 8.2.1. ¿Cuál de estos elementos es más probable que forme iones con una carga de 2+? (a) Li, (b) Ca, (c) O, (d) P, (e) Cl.
Muestra 8.2.2. Prediga las cargas de los iones formados cuando el magnesio reacciona con el nitrógeno.
6.5 Configuración electrónica de un ion en elementos de transición y tierras raras
Debido a que las energías de ionización aumentan rápidamente por cada electrón eliminado, las energías reticulares de los compuestos iónicos son generalmente lo suficientemente grandes como para compensar la pérdida de hasta tres electrones de los átomos. Así, encontramos cationes con cargas de 1+, 2+ o 3+ en compuestos iónicos. La mayoría de los metales de transición, sin embargo, tienen más de tres electrones más allá de un núcleo de gas noble. La plata, por ejemplo, tiene una configuración electrónica (Kr)4d105s1. Los metales del grupo 1B (Cu, Ag, Au) a menudo se presentan como iones 1+ (como en CuBr y AgCl). Al formar Ag+, el electrón 5s se pierde, dejando una subcapa 4d completamente llena. Como en este ejemplo, los metales de transición generalmente no forman iones que tengan una configuración de gas noble. La regla del octeto, aunque útil, tiene un alcance claramente limitado.
Recuerde que cuando se forma un ion positivo a partir de un átomo, los electrones siempre se pierden primero en la subcapa que tiene el mayor valor de número cuántico principal. Por lo tanto, al formar iones, los metales de transición pierden primero los electrones de la capa de valencia y luego tantos electrones d como sean necesarios para alcanzar la carga del ion mas estable. Por ejemplo, al formar Fe2+ a partir de Fe, que tiene la configuración electrónica (Ar)3d 64s2, los dos electrones 4s se pierden, lo que lleva a una configuración (Ar)3d6. La eliminación de un electrón adicional da Fe3+, cuya configuración electrónica es (Ar)3d5.
7. Energía reticular
|| REGRESAR AL INDICE ||
La formación de cloruro de sodio a partir de sodio y cloro es muy exotérmica, como lo indica la gran entalpía negativa del valor de formación ∆Hf° = — 410.9 kJ. Tenga en cuenta que un valor negativo en una entalpía estándar de formación es igual a decir que el calor de reacción se emite. Si analizamos la siguiente tabla de entalpias estándar de formación para compuestos iónicos nos daremos cuenta de que tienen calores de reacción negativos o exotérmicos. ¿Qué factores hacen que la formación de compuestos iónicos sea tan exotérmica?
7.1 La conservación de la energía para iones infinitamente separados
En la Ecuación 6.2 representamos la formación de NaCl como la transferencia de un electrón de Na a Cl. Recuerde que la pérdida de electrones de un átomo es siempre un proceso endotérmico. Retirar un electrón de Na(g) para formar Na+(g), por ejemplo, requiere 496 kJ/mol.
Cuando un no metal gana un electrón, el proceso generalmente es exotérmico, como se ve por las afinidades electrónicas negativas de los elementos. Agregar un electrón a Cl(g), por ejemplo, libera 349 kJ/mol. A partir de las magnitudes de estas energías, podemos ver que la transferencia de un electrón de un átomo de Na a un átomo de Cl no sería exotérmica; el proceso general sería un proceso endotérmico que requiere 496 - 349 = 147 kJ/mol.
Este proceso endotérmico corresponde a la formación de iones de sodio y cloruro que están infinitamente separados; en otras palabras, el cambio de energía positivo supone que los iones no interactúan entre sí, lo cual es bastante diferente de la situación en los sólidos iónicos.
7.2 La conservación de la energía para una matriz iónica
La razón principal por la que los compuestos iónicos son estables es la atracción entre iones de carga opuesta. Esta atracción une a los iones, liberando energía y haciendo que los iones formen una matriz sólida, o red, como la que se muestra:

Figura 7.1. La estructura cristalina del cloruro de sodio. Si no se proporcionara ninguna clave de color, ¿cómo sabría qué bola de color representaba Na+ y cuál representaba Cl-? La respuesta radica en el tamaño relativo de los iones, cuando el cloro absorbe electrones su electronegatividad disminuye y por lo tanto su volumen atómico se expande, por el contrario, cuando el sodio pierde electrones su electronegatividad aumenta y su ion se contrae, de allí que el ion cloruro sea más pequeño que el ion sodio.
Una medida de cuánta estabilización resulta de disponer iones de carga opuesta en un sólido iónico está dada por la energía reticular, que es la energía requerida para separar completamente un mol de un compuesto iónico sólido en sus iones gaseosos.
Para imaginar este proceso para el NaCl, imagine que la estructura de la Figura 7.1 se expande desde adentro, de modo que las distancias entre los iones aumentan hasta que los iones están muy separados. Este proceso requiere 788 kJ/mol, que es el valor de la energía reticular. Note que este proceso es altamente endotérmico. El proceso inverso, la unión de Na+(g) y Cl-(g) para formar NaCl(s), es por lo tanto altamente exotérmico ∆H = -788 kJ/mol.
Observe que, dadas las unidades, esta es una energía reticular estándar, pues está definida para un mol de sustancia, y en consecuencia, dependiendo de la cantidad de sustancia podremos tener diferentes valores. De hecho, otra cantidad de sustancia que es muy común de emplear en este contexto aparte de un mol es una partícula, de allí que tendremos energías de red definidas para una sola partícula iónica. Aunque conceptualmente la diferencia no parece ser muy grande, numéricamente lo sí que lo es, esto se debe a que ambas están separadas por un número de Avogadro de distancia, lo cual nos puede llevar a cometer el error de calcular la energía reticular para una sola partícula y creer que cometimos operaciones incorrectas cuando lo que pasa es que estamos comparando con una tabla que tiene las energías de red, pero ponderadas para un mol de sustancia.
7.3 Energía de una red iónica o energía reticular
Por lo anterior, modificaremos la ecuación que nos da los libros de texto para calcular la energía reticular, en una función que nos permita calcular específicamente la energía reticular estándar o molar, en términos de parámetros que sean indiferentes con respecto a si estamos analizando partículas o moles de sustancia. el parámetro de carga que es indiferente ante esas dos situaciones es la carga relativa z, que normalmente usamos como sinónimo del estado de oxidación.
Con lo que obtenemos:

Figura 7.2. Energía reticular en términos de z1 y z2 que son los números de carga de los iones monoatómicos “normalmente alguno de sus estados de oxidación”; ro es la distancia entre sus centros en metros, y κ3 es una constante de proporcionalidad, 1.39 x 10-7 kJ m / mol.
Tenga en cuenta que, como se ve en la deducción, la constante de proporcionalidad en este sistema de ecuaciones no es la misma en todos los casos, de allí el subíndice que le damos κ3, Dado que existen otras dos versiones de esa obstante. La Eq. 7.1 indica que la interacción de atracción entre dos iones de carga opuesta aumenta a medida que aumentan las magnitudes de sus cargas y disminuye la distancia entre sus centros. Por lo tanto, para una disposición dada de iones, la energía de la red aumenta a medida que aumentan las cargas de los iones y disminuyen sus radios. La variación en la magnitud de las energías de la red depende más de la carga iónica que del radio iónico porque los radios iónicos varían solo en un rango limitado en comparación con las cargas.
7.4 El radio más estable o r0
Como se mencionó anteriormente la separación entre dos iones de una sustancia iónica puede variar dependiendo de las condiciones energéticas del sistema.

Figura 7.3. El efecto de la carga y la distancia sobre la fuerza de las interacciones electrostáticas. A medida que aumenta la carga de los iones o disminuye la distancia entre los iones, también lo hace la fuerza de las interacciones atractivas (−…+) o repulsivas (−…− o +…+). La fuerza de estas interacciones está representada por el grosor de las flechas.
Para iones de carga opuesta, la atracción aumenta a medida que aumenta la carga y disminuye a medida que aumenta la distancia entre los iones.

Figura 7.4. Gráfica de energía potencial (kJ) versus distancia internuclear (pm o 10-12 m) para la interacción entre iones con diferentes cargas de un ion gaseoso de Na+ y un ion gaseoso de Cl−. La energía del sistema alcanza un mínimo a una distancia particular (r0) cuando las interacciones de atracción y repulsión están balanceadas. A continuación se muestra una aplicación de pHet que ilustra el mismo punto para los átomos neutros. Puede mover el átomo no anclado con respecto al anclado arrastrándolo y puede ver en qué parte de la curva de potencial se encuentra en función de la distancia entre ellos. La principal diferencia entre las curvas para la atracción iónica y los átomos neutros es que la fuerza entre los iones es mucho más fuerte y, por lo tanto, la profundidad del pozo es mucho más profunda. Revisaremos esta aplicación cuando hablemos de enlaces que no son iónicos.
Como muestra la curva verde en la mitad inferior de la Figura 7.3, predice que la energía máxima se libera cuando los iones están infinitamente cerca uno del otro, en r = 0. Debido a que los iones ocupan espacio y tienen una estructura con el núcleo positivo siendo rodeados de electrones, sin embargo, no pueden estar infinitamente juntos. A distancias muy cortas, las interacciones repulsivas electrón-electrón entre electrones en iones adyacentes se vuelven más fuertes que las interacciones atractivas entre iones con cargas opuestas, como lo muestra la curva roja en la mitad superior de la Figura 7.3. La energía total del sistema es un equilibrio entre las interacciones atractivas y repulsivas. La curva morada de la figura 7.3 muestra que la energía total del sistema alcanza un mínimo en r0, el punto donde las repulsiones y atracciones electrostáticas están exactamente equilibradas. Esta distancia es la misma que la distancia de enlace medida experimentalmente.
LibreChem
Sin embargo, hasta ese punto hemos obtenido valores contradictorios de la energía reticular para el cloruro de sodio en 2 fuentes diferentes. y eso se debe a que la ecuación 7.1 es un valor teórico, mientras que el valor dado por el libro de texto de Brown es un valor medido experimentalmente, lo cual implica necesariamente que la ecuación 7.1 está incompleta. la razón de esa incompletitud radica en que los iones no sólo forman redes, sino que dichas redes tienen características propias dependiendo de las interacciones de los elementos que no son predecibles fácilmente, de allí que habrá diferencias entre los valores esperados y medidos.
7.5 Radio desconocido
Algunas variantes de ejercicios de lápiz y papel para estas situaciones incluyen análisis cualitativos donde nos ofrecen el valor de la distancia entre los iones. Para estos casos deberemos emplear el radio iónico de los elementos involucrados.

Figura 7.5. Radios de los iones más comunes de algunos elementos de la tabla periódica. tenga en cuenta que la tendencia periódica para el radio es opuesta que la de la electronegatividad, esto se debe a que cuando un elemento muy electronegativo absorbe electrones pierde electronegatividad y por lo tanto su nube electrónica se distiende debido a una menor atracción de electrones hacia el núcleo aumentando su radio.
Teniendo en cuenta los elementos de un mismo grupo columna aquellos que tienen un mayor número atómico también tendrán un mayor radio iónico. Adicionalmente deberemos asumir que entre mayor sea el radio iónico mayor será la separación entre los guiones en la red iónica.

Distancia de separación entre los iones en la red como la suma de los radios iónicos.
Dado lo anterior dado que r0 se encuentra en el denominador, entre más grande es el ion, más pequeña es la energía reticular y viceversa. Por lo tanto, si no nos dan valores numéricos el algoritmo de solución es el siguiente:
👉 estimar primero el producto de números de carga relativo (estados de oxidación), pues este término afecta más profundamente a la energía reticular que cualquier otro. Entre mayor sea el producto mayor será la energía reticular.
👉 si el producto de números de carga relativa es igual, entonces indagamos la suma de radios iónicos.
👉 si no tenemos acceso a los valores numéricos de los radios iónicos, determinamos la posición relativa en la columna de la tabla periódica. Normalmente en ejercicios de lápiz y papel las sustancias a comparar deberán permitir un análisis sencillo en términos de una columna, entre más abajo se encuentra un elemento en su columna, su radio iónico va a ser más grande.
Química la ciencia central
Muestra 8.1. Ordene los compuestos iónicos NaF, CsI y CaO en orden creciente de energía reticular.
Práctica 8.1.2. ¿Qué sustancia espera que tenga la mayor energía reticular: MgF2, CaF2 o ZrO2?
8. Enlace covalente y su representación
|| REGRESAR AL INDICE ||
La gran mayoría de las sustancias químicas no tienen las características de los materiales iónicos. La mayoría de las sustancias con las que entramos en contacto a diario, como el agua, suelen ser gases, líquidos o sólidos con puntos de fusión y ebullición bajos. Muchos, como la gasolina, se vaporizan fácilmente. Muchos son flexibles en sus formas sólidas, por ejemplo, bolsas de plástico y cera.
Para la gran clase de sustancias que no se comportan como sustancias iónicas, necesitamos un modelo diferente para describir el enlace entre los átomos. G. N. Lewis razonó que los átomos podrían adquirir una configuración electrónica de gas noble al compartir electrones con otros átomos. Un enlace químico formado al compartir un par de electrones es un enlace covalente.
La molécula de hidrógeno, H2, proporciona el ejemplo más simple de un enlace covalente. Los átomos de H2 se mantienen unidos principalmente porque los dos núcleos positivos son atraídos por la concentración de carga negativa entre ellos. En esencia, el par de electrones compartidos en cualquier enlace covalente actúa como una especie de "pegamento / puente" para unir los átomos.
8.1 Estructuras de Lewis
Normalmente emplearemos las estructuras de Lewis para modelar el enlace covalente, donde un par de electrones van a funcionar como el puente electrónico entre los dos átomos. La clave era aquí radica en que los dos electrones que constituyen el puente van a funcionar como electrones de valencia para los dos átomos unidos. Así por ejemplo, para explicar la molécula de cloro elemental que tiene dos átomos de cloro unidos Cl2, cada átomo de cloro compromete un electrón en el puente covalente, de forma tal que los dos electrones que forman el enlace cuentan para ambos átomos, lo que nos obliga a contar ocho electrones de valencia en cada cloro.

Figura 8.1. Representación por estructura de Lewis de la molécula de cloro elemental. A la izquierda a la forma de punto y enlace; y a la derecha la forma de guion y enlace.
Los pares de electrones no enlazantes que quedan dibujados alrededor de los átomos son importantes debido a que generan cargas electrostáticas negativas que pueden atraer a estructuras positivas a su alrededor débilmente.
Para los no metales, el número de electrones de valencia en un átomo neutro es el mismo que el número de grupo que emplean la nomenclatura CAS desde IA hasta VIIIA. Por lo tanto, uno podría predecir que los elementos VIIA, como flúor, formarían un enlace covalente para lograr un octeto; Los elementos VIA, como el O, formarían dos enlaces covalentes; Los elementos VA, como N, formarían tres; y los elementos IVA, como C, formarían cuatro. Estas predicciones se confirman en muchos compuestos, como en, por ejemplo, los compuestos con hidrógeno de los no metales de la segunda fila de la tabla periódica.

Figura 8.2. Representación por estructura de punto y enlace Lewis de las moléculas de HF, H2O, NH3, y CH4.
8.2 Enlaces múltiples
Un par de electrones compartidos constituye un enlace covalente simple, generalmente denominado simplemente enlace simple. En muchas moléculas, los átomos alcanzan octetos completos al compartir más de un par de electrones. Cuando dos pares de electrones son compartidos por dos átomos, se dibujan dos líneas en la estructura de Lewis, que representan un doble enlace. En el dióxido de carbono, por ejemplo, el enlace se produce entre el carbono, con cuatro electrones de valencia, y el oxígeno, con seis:

Figura 8.3. Representación por estructura de punto y enlace Lewis para la formación de la molécula de CO2.
Como muestra el diagrama, cada átomo de oxígeno adquiere un octeto al compartir dos pares de electrones con el carbono. En el caso del CO2, el carbono adquiere un octeto al compartir dos pares de electrones con cada uno de los dos átomos de oxígeno; cada doble enlace implica cuatro electrones. Un enlace triple corresponde a la compartición de tres pares de electrones, como en la molécula N2:

Figura 8.4. Representación por estructura de punto y enlace Lewis para la formación de la molécula de N2.
Debido a que cada átomo de nitrógeno tiene cinco electrones de valencia, se deben compartir tres pares de electrones para lograr la configuración del octeto. Las propiedades del N2 están en completo acuerdo con su estructura de Lewis. El nitrógeno es un gas diatómico con una reactividad excepcionalmente baja que resulta del enlace nitrógeno-nitrógeno muy estable. Los átomos de nitrógeno están separados por solo 1.10 Å. La corta distancia de separación entre los dos átomos de N es el resultado del triple enlace entre los átomos. Como regla general, la longitud del enlace entre dos átomos disminuye a medida que aumenta el número de pares de electrones compartidos.
8.3 Muchas excepciones al octeto
Ese modelo es muy bonito para los átomos de los grupos representativos, es decir los del grupo IA hasta VIII A, pero no es muy bueno para representar los elementos que se encuentran en los metales de transición y tierras raras, o cuando algunos elementos de los grupos representativos hibridan y cambian su estado de oxidación para tener un poder de enlace diferente del que se produce con la regla del octeto.
9. Enlace covalente polar y electronegatividad
|| REGRESAR AL INDICE ||
Cuando dos átomos idénticos se unen, como en Cl2 o H2, los pares de electrones deben compartirse por igual. Cuando dos átomos de lados opuestos de la tabla periódica se unen, como el NaCl, los electrones tienden a permanecer más cerca del elemento con mayor electronegatividad, la unión por lo tanto no es por un puente de electrones llamado enlace covalente, sino por la atracción electrostática de cargas opuestas. Los enlaces que se encuentran en la mayoría de las sustancias se encuentran en algún lugar entre estos extremos, es decir donde los electrones del enlace covalente tenderán a permanecer más tiempo cerca del elemento más electronegativo pero sin que podamos hablar de que se generen propiedades iónicas verdaderas en las sustancias macroscópicas.
La polaridad del enlace es una medida de cuán equitativa o desigualmente se comparten los electrones en cualquier enlace covalente. Un enlace covalente no polar es aquel en el que los electrones se comparten por igual, como en Cl2 y N2. En un enlace covalente polar, uno de los átomos ejerce una mayor atracción por los electrones de enlace que el otro. Si la diferencia en la capacidad relativa para atraer electrones es lo suficientemente grande, se forma un enlace iónico. Tenga muy en cuenta que la noción de enlace covalente o iónico dependerá de las propiedades que exhibe la sustancia macroscópica más que cálculos numéricos que realicemos posteriormente, y aunque los cálculos nos permitirán predecir muchas sustancias, tendremos excepciones.
9.1 Electronegatividad
Usamos una cantidad llamada electronegatividad “símbolo chi χ según el libro de oro de la IUPAC” para estimar si un enlace dado es covalente no polar, covalente polar o iónico. La electronegatividad se define como la capacidad de un átomo en una molécula para atraer electrones hacia sí mismo. Cuanto mayor es la electronegatividad de un átomo, mayor es su capacidad para atraer electrones hacia sí mismo. La electronegatividad de un átomo en una molécula está relacionada con la energía de ionización del átomo y la afinidad electrónica, que son propiedades de los átomos aislados. Un átomo con una afinidad electrónica muy negativa y una energía de ionización alta atrae electrones de otros átomos y se resiste a que sus electrones sean atraídos; por lo tanto es altamente electronegativo.
Los valores de electronegatividad se pueden basar en una variedad de propiedades, no solo en la energía de ionización y la afinidad electrónica. El químico estadounidense Linus Pauling (1901–1994) desarrolló la primera y más utilizada escala de electronegatividad, que se basa en datos termoquímicos. Generalmente hay un aumento en la electronegatividad de izquierda a derecha a lo largo de un período, es decir, desde los elementos más metálicos hasta los más no metálicos.

Figura 9.1. Aumento de la electronegatividad en la tabla periódica empleando la escala Pauling. En la imagen podemos ver claramente que el elemento de mayor electronegatividad es el flúor el elemento de menor electronegatividad es el francio, en consecuencia la diferencia de electronegatividad de fluoruro de francio deberá ser la más alta y por lo tanto la más iónica.
Con algunas excepciones (especialmente en los metales de transición), la electronegatividad disminuye al aumentar el número atómico en un grupo. Esto es lo que esperamos porque sabemos que las energías de ionización disminuyen al aumentar el número atómico en un grupo y las afinidades electrónicas no cambian mucho.
9.2 Polaridad de un enlace
Un enlace polar también denominado covalente polar es un tipo de enlace covalente que se encuentra a medio camino entre el enlace covalente perfecto y el enlace iónico perfecto, para estimar la existencia del enlace covalente polar generalmente realizamos la diferencia de electronegatividad es de una pareja de elementos que forman un enlace químico.

Valor absoluto de la diferencia de electronegatividad es, en términos de las electronegatividades del elemento uno (I) y el elemento 2 (II).
Química la ciencia central
Muestra 8.4a. ¿Cuál enlace es más polar? B - Cl o C - Cl. Indique en cada caso qué átomo tiene la carga negativa parcial.
Muestra 8.4b. ¿Cuál enlace es más polar? P - F o P - Cl. Indique en cada caso qué átomo tiene la carga negativa parcial.
Práctica 8.4.1. ¿Cuál de los siguientes enlaces es el más polar? (a) H-F, (b) H-I, (c) Se-F, (d) N-P, (e) Ga-Cl.
Práctica 8.4.2. ¿Cuál de los siguientes enlaces es más polar: S-Cl, S-Br, Se-Cl o Se-Br?
Para enlaces entre átomos de un mismo elemento como sucede en las moléculas diatómicas tales como F2, Cl2, H2, o como sucede en estructuras complejas tales como las de los hidrocarburos H3C—CH3, la electronegatividad de los dos átomos es igual porque pertenecen al mismo elemento, de allí que la diferencia de electronegatividades será cero, lo cual define al enlace covalente perfecto, en consecuencia entre más cercano a cero o sea la diferencia de electronegatividad es en su valor absoluto, más covalente será el enlace.
Para una sustancia como el fluoruro de litio LiF o el fluoruro de francio FrF, la diferencia de electronegatividad es es muy alta, la mayor diferencia de electronegatividad es la encontramos en el FrF (|Δχ| = 4.0 – 0.7 = 3.3), aunque la diferencia de electronegatividad es de LiF va a ser muy cercana (|Δχ| = 4.0 – 1.0 = 3.0), y en consecuencia entre más cercana sea el valor absoluto de la diferencia de electronegatividad es a 3.3, más iónico será ese enlace.
En HF, el átomo de flúor tiene una electronegatividad mayor que el átomo de hidrógeno, con el resultado de que los electrones se comparten de manera desigual: (|Δχ| = 4.0 – 2.1 = 1.9) el enlace es polar. En general, un enlace covalente polar resulta cuando los átomos difieren en electronegatividad. En HF, el átomo de flúor más electronegativo atrae la densidad de electrones lejos del átomo de hidrógeno menos electronegativo, dejando una carga positiva parcial en el átomo de hidrógeno y una carga negativa parcial en el átomo de flúor. Podemos representar esta distribución de carga parcial empleando la letra griega delta más el signo de polaridad pertinente como:

Figura 9.2. Modelo que representa las cargas relativas en una molécula covalente polar. El δ+ y δ- (léase “delta más” y “delta menos”) simbolizan las cargas positivas y negativas parciales, respectivamente.
En un enlace polar, estos números son menores que las cargas completas de los iones, pero serán lo suficientemente grandes como para generar atracciones y repulsiones en sustancias y afectar a las propiedades físicas de estas. La pregunta ahora es cuál es la frontera entre el iónico y covalente polar, y la respuesta será muy difícil de responder debido a las excepciones, por lo general he visto que la frontera que lo separa se encuentra en el rango de entre 1.7 a 2.1.
9.3 Momento dipolar
La diferencia de electronegatividad entre H y F conduce a un enlace covalente polar en la molécula de HF. Como consecuencia, hay una concentración de carga negativa en el átomo de F más electronegativo, dejando al átomo de H menos electronegativo en el extremo positivo de la molécula. Una molécula como HF, en la que los centros de carga positiva y negativa no coinciden, es una molécula polar, que funciona como un pequeño imán. Por lo tanto, describimos tanto los enlaces como las moléculas enteras como polares y no polares. Podemos indicar la polaridad de la molécula de HF de dos maneras:

Figura 9.3. Modelos que representan el momento dipolar en una molécula. En la notación de la derecha, la flecha indica el desplazamiento de la densidad electrónica hacia el átomo de flúor. El extremo cruzado de la flecha se puede considerar como un signo más que designa el extremo positivo de la molécula.
La polaridad ayuda a determinar muchas propiedades que observamos a nivel macroscópico en el laboratorio y en la vida cotidiana. Las moléculas polares se alinean entre sí, atrayéndose el extremo negativo de una molécula y el extremo positivo de otra. Las moléculas polares también son atraídas por los iones. El extremo negativo de una molécula polar es atraído por un ion positivo y el extremo positivo es atraído por un ion negativo.
Estas interacciones explican muchas propiedades de líquidos, sólidos y soluciones. La separación de carga dentro de las moléculas juega un papel importante en los procesos de conversión de energía como la fotosíntesis.
¿Cómo podemos cuantificar la polaridad de una molécula? Siempre que dos cargas eléctricas de igual magnitud pero de signo contrario están separadas por una distancia, se establece un dipolo. La medida cuantitativa de la magnitud de un dipolo se llama su momento dipolar, denotado con la letra griega mu, μ. Si dos cargas iguales y opuestas Q+ y Q- están separadas por una distancia r, la magnitud del momento dipolar es el producto de Q y r:

Magnitud del momento dipolar en términos de la carga y la distancia. Esta expresión nos dice que el momento dipolar aumenta a medida que aumenta la magnitud de Q y cuando aumenta r. Cuanto mayor sea el momento dipolar, más polar será el enlace. Para una molécula no polar, como F2, el momento dipolar es cero porque no hay separación de carga.
Los momentos dipolares se pueden medir experimentalmente y generalmente se expresan en debyes (D), una unidad que equivale a 3.34 x 10-30 C-m (coulomb-metros). Para las moléculas, normalmente medimos la carga en unidades de la carga electrónica elemental 1.60 x 10-19 C, y la distancia en angstroms.
Brown13 química la ciencia central
La tabla 8.3 presenta las longitudes de enlace y los momentos dipolares de los haluros de hidrógeno. Observe que a medida que avanzamos de HF a HI, la diferencia de electronegatividad disminuye y la longitud del enlace aumenta. El primer efecto disminuye la cantidad de carga separada y hace que el momento dipolar disminuya de HF a HI, aunque la longitud del enlace aumenta. Cálculos idénticos a los utilizados en el ejercicio de Muestra 8.5 muestran que las cargas de los átomos disminuyen de 0.41+ y 0.41- en HF a 0.057+ y 0.057- en HI. Podemos visualizar el grado variable de cambio de carga electrónica en estas sustancias a partir de representaciones generadas por computadora basadas en cálculos de distribución de electrones, como se muestra en la Figura 8.10. Para estas moléculas, el cambio en la diferencia de electronegatividad tiene un mayor efecto sobre el momento dipolar que el cambio en la longitud del enlace.

Figura 9.4. Separación de carga en los haluros de hidrógeno. En HF, el F fuertemente electronegativo aleja gran parte de la densidad electrónica de H. En HI, el I, al ser mucho menos electronegativo que F, no atrae los electrones compartidos con tanta fuerza y, en consecuencia, hay mucha menos polarización de los electrones.
Antes de dejar esta sección, volvamos a la molécula LiF de la figura anterior. En condiciones estándar, el LiF existe como un sólido iónico con una disposición de átomos análoga a la estructura del cloruro de sodio. Sin embargo, es posible generar moléculas de LiF vaporizando el sólido iónico a alta temperatura. Las moléculas tienen un momento dipolar de 6.28 D y una distancia de enlace de 1.53 Å. A partir de estos valores podemos calcular la carga del litio y el flúor en 0.857+ y 0.857-, respectivamente. Este enlace es extremadamente polar y la presencia de cargas tan grandes favorece fuertemente la formación de una red iónica extendida en la que cada ion de litio está rodeado por iones de fluoruro y viceversa. Pero incluso aquí, las cargas determinadas experimentalmente en los iones todavía no son 1+ y 1-. Esto nos dice que incluso en los compuestos iónicos, todavía hay alguna contribución covalente al enlace.
10. Diferenciando el enlace iónico del enlace covalente
|| REGRESAR AL INDICE ||
Para comprender las interacciones responsables del enlace químico, es ventajoso tratar el enlace iónico y el covalente por separado. Ese es el enfoque adoptado en este capítulo, así como en la mayoría de los demás textos de química de nivel universitario. En realidad, sin embargo, existe un continuo entre los extremos del enlace iónico y covalente. Esta falta de una separación bien definida entre los dos tipos de vínculos puede parecer inquietante o confusa al principio.
Los modelos simples de enlaces iónicos y covalentes presentados en este capítulo contribuyen en gran medida a comprender y predecir las estructuras y propiedades de los compuestos químicos. Cuando el enlace covalente es dominante, esperamos que los compuestos existan como sustancias moleculares, que tengan todas las propiedades que asociamos con las sustancias moleculares, como puntos de fusión y ebullición relativamente bajos y un comportamiento no electrolítico cuando se disuelven en agua. Cuando el enlace iónico es dominante, esperamos que los compuestos sean sólidos quebradizos, de alto punto de fusión con estructuras reticulares extendidas, que exhiben un fuerte comportamiento electrolítico cuando se disuelven en agua.
Naturalmente, hay excepciones a estas caracterizaciones generales. No obstante, la capacidad de categorizar rápidamente las interacciones de enlace predominantes en una sustancia como covalente o iónica brinda una comprensión considerable de las propiedades de muchas sustancias, en otras palabras, con estas generalizaciones es más probable que juzguemos bien a la mayoría que si se ajusta. La pregunta entonces se convierte en la mejor manera de reconocer qué tipo de vínculo domina.
El enfoque más simple es asumir que la interacción entre un metal y un no metal es iónica y que entre dos no metales es covalente. Si bien este esquema de clasificación es razonablemente predictivo, hay demasiadas excepciones para usarlo a ciegas. Por ejemplo, el estaño es un metal y el cloro es un no metal, pero el SnCl4 es una sustancia molecular que existe como un líquido incoloro a temperatura ambiente. Se congela a -33 °C y hierve a 114 °C. Las características del SnCl4 no son las típicas de una sustancia iónica. ¿Existe una forma más predecible de determinar qué tipo de enlace prevalece en un compuesto?
Un enfoque más sofisticado consiste en utilizar la diferencia de electronegatividad como criterio principal para determinar si predominarán los enlaces iónicos o covalentes. Este enfoque predice correctamente que el enlace en SnCl4 será covalente polar en función de una diferencia de electronegatividad de 1.2 y, al mismo tiempo, predice correctamente que el enlace en NaCl será predominantemente iónico en función de una diferencia de electronegatividad de 2.1.
La evaluación de enlaces basada en la diferencia de electronegatividad es un sistema útil, pero tiene un inconveniente. Los valores de electronegatividad dados en la mayoría de tablas periódicas no tienen en cuenta los cambios en los enlaces que acompañan a los cambios en el estado de oxidación del metal. Por ejemplo, la diferencia de electronegatividad entre el manganeso y el oxígeno como |Δχ(MgO)| = 3.5 – 1.5 = 2.0, que cae en el rango donde el enlace normalmente se considera iónico (la diferencia de electronegatividad para NaCl es Δχ(NaCl)| = 3.0 – 0.9 = 2.1). Por lo tanto, no sorprende saber que el óxido de manganeso (II), MnO, es un sólido verde que se funde a 1842 °C y tiene la misma estructura cristalina que el NaCl.
Sin embargo, el enlace entre el manganeso y el oxígeno no siempre es iónico. El óxido de manganeso (VII), Mn2O7, es un líquido verde que se congela a 5.9 °C, lo que indica que predominan los enlaces covalentes en lugar de los iónicos. El cambio en el estado de oxidación del manganeso es responsable del cambio en la unión. En general, a medida que aumenta el estado de oxidación de un metal, también aumenta el grado de enlace covalente, o dicho de otra manera, aumenta su naturaleza no metálica. Cuando el estado de oxidación del metal es muy positivo (en términos generales, +4 o más), podemos esperar una covalencia significativa en los enlaces que forma con los no metales, y por ende tenderá a comportarse como no metales, generando por ejemplo aniones ácidos con oxígeno. Así, los metales en altos estados de oxidación forman sustancias moleculares, como Mn2O7, o aniones poliatómicos, como MnO4- y CrO42-, en lugar de compuestos iónicos.
11. Dibujando estructuras de Lewis
|| REGRESAR AL INDICE ||
Las estructuras de Lewis, también conocidas como diagramas de puntos de Lewis, muestran la relación de enlace entre los átomos de una molécula y los pares de electrones solitarios en la molécula. Las estructuras de Lewis también pueden ser útiles para predecir la geometría molecular junto con los orbitales híbridos. Un compuesto puede tener múltiples formas de resonancia que también son todas estructuras de Lewis correctas. Esta sección discutirá las reglas para escribir correctamente las estructuras de Lewis.
Escribir estructuras de Lewis puede ser a veces complicado y algo difícil. Un compuesto puede tener varias estructuras de Lewis que contribuyen a la forma del compuesto en general, por lo que una estructura de Lewis de un compuesto puede no ser necesariamente exactamente como se ve el compuesto. Pero antes de comenzar, hay algunas cosas que debe saber.
👉 Un electrón se representa como un punto.
👉 Un enlace, que está formado por 2 electrones compartidos, se representa mediante dos puntos entre los átomos enlazados o una línea (líneas de estructuras de Kekulé).
👉 Los enlaces dobles y los enlaces triples se representan como dos y tres líneas/(pares de electrones), respectivamente.
👉 Los pares solitarios en los bordes exteriores de un átomo se representan como dos puntos.
👉 Los electrones representados en una estructura de Lewis son los electrones de la capa externa, que se denominan electrones de valencia. Esto se debe a que son los que participan en las reacciones químicas.
Los ejemplos enumerados a continuación pueden o no ser consistentes para cada paso con el fin de mostrar múltiples ejemplos, ya que todos los pasos se aplican a cada compuesto, y algunos pasos cambian dependiendo del tipo de compuesto que estemos trabajo. Ahora estamos listos para comenzar.
11.1 Compuestos binarios covalentes
Lo primero es identificar que estemos modelando un compuesto iónico o un compuesto covalente, esto deberíamos hacerlo ya sea identificando que los elementos sean uniones de metal o no metal (iónicos) siempre y cuando se encuentren alejados en la tabla periódica o uniones entre no metales (covalentes). Si los dos elementos metal y no metal se encuentran muy próximos en la tabla periódica, resultará conveniente que realice la diferencia de electronegatividades que vimos en secciones anteriores, para estar seguros de que se trata de un enlace iónico covalente.
Para un compuesto molecular de enlaces covalentes binario ejecutaremos las siguientes reglas.
👉 Identificar el grupo al que pertenece cada elemento.
👉 Determinar cuántos electrones requiere para completar su octeto con enlaces covalentes.
👉 Quien requiere más enlaces será el átomo central que se dibuja con sus electrones de valencia.
👉 Adicionar cuantos átomos sean necesarios para completar los enlaces requeridos para completar el octeto.
👉 En ocasiones debe agregar enlaces múltiples para completar los octetos de átomos.
👉 Sume los electrones de valencia de los átomos separados y compárelos con los de la estructura de Lewis para ver que no se cometieran errores.
A parte de estas reglas generales, tenga en cuenta las siguientes indicaciones
Usa la tabla periódica para ayudarte a determinar el número de electrones de valencia en cada átomo. Para un anión, agregue un electrón al total por cada carga negativa. Para un catión, reste un electrón del total por cada carga positiva. No se preocupe por hacer un seguimiento de qué electrones provienen de qué átomos. Sólo el número total es importante, debido a que en los iones las cargas pueden resonar entre varios átomos y la cosa puede volverse complicada.
Las fórmulas químicas a menudo se escriben en el orden en que los átomos están conectados en la molécula o ion. La fórmula HCN, por ejemplo, indica que el átomo de carbono está unido al H y al N. En muchas moléculas e iones poliatómicos, el átomo central suele escribirse primero, como en CO32 y SF4. Recuerde que el átomo central es generalmente menos electronegativo que los átomos que lo rodean. En otros casos, es posible que necesite más información antes de poder dibujar la estructura de Lewis.
Tenga en cuenta que un átomo de hidrógeno tiene solo un par de electrones a su alrededor.
Química la ciencia central
Muestra 8.6. Dibuje la estructura de Lewis para el tricloruro de fósforo, PCl3.
Práctica 8.6.2. (a) ¿Cuántos electrones de valencia deberían aparecer en la estructura de Lewis para CH2Cl2? (b) Dibuje la estructura de Lewis.
Muestra 8.7. Dibujar la estructura de Lewis para HCN
Práctica 8.7.2. Dibuje la estructura de Lewis para (a) ion NO+, (b) C2H4.
Para los iones debemos olvidarnos de parte de las reglas que mencionamos anteriormente, en su lugar lo que vamos a hacer es:
👉 calcular la cantidad total de electrones de valencia que suman todos los átomos.
👉 sumar o restar la carga del ion
👉 dibujar el átomo central y los átomos periféricos
👉 conectar el átomo central y los átomos periféricos con un solo enlace
👉 completar el octeto del átomo central con electrones no enlazantes que hagan falta
👉 completar los objetos de los átomos periféricos con los electrones no enlazantes que hagan falta
👉 sumar los electrones enlazantes y no enlazantes que empleamos en nuestra hipótesis y
👉 compararla con el cálculo inicial, si el resultado es la misma cantidad de electrones, entonces la estructura de leguis será satisfactoria.
Química la ciencia central
Muestra 8.8. Dibuje la estructura de Lewis para el ion BrO3-.
Práctica 8.8.1. ¿Cuántos pares de electrones no enlazantes hay en la estructura de Lewis del ion peróxido, O22-? (a) 7, (b) 6, (c) 5, (d) 4, (e) 3.
Práctica 8.8.2. Dibuje la estructura de Lewis para (a) ClO2-, (b) PO43- .
11.2 Compuestos del tipo HaXOb
Estos son conocidos como los ácidos oxácidos, lo que tienen de característico al intentar las hacer el dibujo de su estructura del Lewis es que los hidrógenos no van a estar unidos al átomo central, por el contrario, van a estar unidos a átomos periféricos de oxígeno. en consecuencia, lo que vamos a hacer en estos casos es:
👉 dibujar primero el átomo central
👉 posteriormente los oxígenos periféricos
👉 unir con un enlace simple del átomo central y los enlaces periféricos
👉 dibujar un hidrógeno adyacente a un oxígeno lo más separado que podamos
👉 dibujar un enlace simple entre oxígeno e hidrógeno
👉 completar los octetos
👉 revisar que la suma de electrones de valencia concuerde con los electrones que hemos empleado para la estructura que dibujamos
También debemos tener en cuenta que ese tipo de moléculas puede tener más de una estructura de Lewis permisible, de hecho, existen otras estructuras que emplean el concepto de octeto extendido. Según las reglas para dibujar estructuras de Lewis, ambas estructuras tienen características indeseables. Sin embargo, los cálculos de la estructura electrónica de la mecánica cuántica encuentran poca participación del orbital d en el enlace, como se esperaría para el enlace de octetos expandidos, lo que sugiere que la estructura que podemos dibujar aplicando las reglas básicas es más adecuada. Por lo tanto, generalmente no hay razón para dibujar estructuras con octetos expandidos, porque normalmente no contribuyen significativamente a la estructura general de la molécula.
Ejemplo.Dibujar la estructura de Lewis sin octeto expandido para H2SO4.
Ejemplo.Dibujar la estructura de Lewis sin octeto expandido para H3PO4.
11.3 Compuestos binarios iónicos
👉 Identificar el grupo al que pertenece cada elemento.
👉 Determinar el ion más probable de cada elemento. Usar el estado de oxidación para metales de transición y tierras raras, y el número de grupo para metales representativos.
👉 Ejecutar la ley de la conservación de la carga para cuantificar el número de átomos de cada elemento.
👉 Consignar el número de iones como si fueran coeficientes algebraicos, mientras que los iones de cada elemento quedan encerrados entre corchetes.
👉 los iones positivos o cationes no portan electrones al interior de su corchete.
👉 los iones negativos o aniones deberán tener su octeto completo.
👉 la cantidad de electrones que pierden todos los cationes debe ser igual a la cantidad de electrones que ganan todos los aniones.
Ejemplo.Dibujar la estructura de Lewis del NaCl
Ejemplo.Dibujar la estructura de Lewis del CaO
Ejemplo.Dibujar la estructura de Lewis del Ca3N2
Ejemplo.Dibujar la estructura de Lewis para Fe2O3
11.4 Cargas formales
Cuando dibujamos una estructura de Lewis, estamos describiendo cómo se distribuyen los electrones en una molécula o ion poliatómico. En algunos casos, podemos dibujar dos o más estructuras de Lewis válidas para una molécula que obedecen todas la regla del octeto. Se puede pensar que todas estas estructuras contribuyen a la disposición real de los electrones en la molécula, pero no todas contribuirán en la misma medida. ¿Cómo decidimos cuál de varias estructuras de Lewis es la más importante? Un enfoque es hacer algo de "contabilidad" de los electrones de valencia para determinar la carga formal de cada átomo en cada estructura de Lewis. La carga formal de cualquier átomo en una molécula es la carga que tendría el átomo si cada par de electrones de enlace en la molécula se compartiera por igual entre sus dos átomos. Para calcular la carga formal de cualquier átomo en una estructura de Lewis, asignamos electrones al átomo de la siguiente manera:
👉 El número de electrones no enlazantes del elemento I-ésimo foco del análisis Ne(I).
👉 El número de enlaces del elemento I-ésimo foco de análisis Nb(I).
👉 Los electrones de valencia del elemento I-ésimo foco de análisis se consigna como NV(I)
👉 La carga formal zF(I) de cada átomo I-ésimo se calcula restando el número de electrones asignados al átomo del número de electrones de valencia en el átomo neutro:
👉 La suma de cargas formales de los elementos involucrados debe ser igual a la carga neta de la molécula en cuestión:

Eq. 11.1. Carga formal zF(J) para un elemento J-cualquiera en términos del número de electrones de valencia NV(J) , el número de enlaces Nb(J) y el número de electrones no enlazantes ze(I) de dicho elemento. Eq. 11.2. Carga de una molécula zj en términos de la suma de cargas formales de los elementos constitutivos zF(J).
Tenga en cuenta que la ecuación anterior debe hacerse átomo por átomo, ya que algunas estructuras de Lewis permiten una carga formal diferentes para átomos de un mismo elemento, por ende, no podemos emplear el subíndice como factor común que acelera el cálculo de la conservación de la carga.
Sin embargo, aun agregando la segunda condición, es posible dibujar mas de una estructura de Lewis, para tales casos seguimos las siguientes recomendaciones extra.
👉 La estructura de Lewis dominante es generalmente aquella en la que los átomos tienen cargas formales más cercanas a cero.
👉 Una estructura de Lewis en la que residen cargas negativas en los átomos más electronegativos suele ser más dominante que una que tiene cargas negativas en los átomos menos electronegativos.
Aunque el concepto de carga formal nos ayuda a organizar estructuras de Lewis alternativas en orden de importancia, es importante que recuerde que las cargas formales no representan cargas reales en los átomos. Estas cargas son solo una convención de contabilidad, una ficción útil. Las distribuciones de carga reales en moléculas e iones no están determinadas por las cargas formales, sino por una serie de otros factores, incluidas las diferencias de electronegatividad entre los átomos. Adicionalmente, para ciertas sustancias con varias estructuras de Lewis posibles, resulta mas conveniente emplear la notación de resonancia para evitar rompernos la cabeza en tratar de identificar la estructura de Lewis teóricamente dominante.
Química la ciencia central
Muestra 8.9. Tres posibles estructuras de Lewis para el ion tiocianato, NCS-, estan dadas en la figura adjunta. Determinar cual es la estructura de Lewis dominante. (pulse en la figura)
12. Estados de oxidación, cargas formales y cargas reales
|| REGRESAR AL INDICE ||
12.1 Números de oxidación o estados de oxidación
En química, el estado de oxidación, o número de oxidación, es la carga hipotética de un átomo si todos sus enlaces a diferentes átomos fueran completamente iónicos. Es decir, al determinar el número de oxidación, todos los electrones compartidos se cuentan con el átomo más electronegativo. Por ejemplo, considere la estructura de Lewis del HCl. Para asignar números de oxidación, ambos electrones en el enlace covalente entre los átomos se asignan al átomo de Cl más electronegativo. Este procedimiento le da al Cl ocho electrones de valencia, uno más que en el átomo neutro. Por lo tanto, su número de oxidación es -1. El hidrógeno no tiene electrones de valencia cuando se cuentan de esta manera, lo que le da un número de oxidación de +1, sin embargo, esto no puede ser real ya que la diferencia de electronegatividades entre los dos núcleos no es tan alta para ser un enlace fuertemente iónico.
El OS describe el grado de oxidación (pérdida de electrones) de un átomo en un compuesto químico. Se simboliza como OSI aunque podemos usarlo como sinónimo de carga relativa de un átomo zI. Conceptualmente, el estado de oxidación puede ser positivo, negativo o cero. Si bien los enlaces completamente iónicos no se encuentran en la naturaleza, muchos enlaces exhiben una fuerte ionicidad, lo que hace que el estado de oxidación sea un predictor útil de la carga (OSI ≈ zI).
El estado de oxidación de un átomo no representa la carga formal "real" de ese átomo, ni ninguna otra propiedad atómica real. Esto es particularmente cierto en los estados de oxidación altos, donde la energía de ionización requerida para producir un ion positivo múltiple es mucho mayor que las energías disponibles en las reacciones químicas. Además, los estados de oxidación de los átomos en un compuesto dado pueden variar según la elección de la escala de electronegatividad utilizada en su cálculo. Así, el estado de oxidación de un átomo en un compuesto es puramente un formalismo o ficción útil. Sin embargo, es importante para comprender las convenciones de nomenclatura de los compuestos inorgánicos. Además, varias observaciones con respecto a las reacciones químicas pueden explicarse a un nivel básico en términos de estados de oxidación.
Los estados de oxidación normalmente se representan mediante números enteros que pueden ser positivos, cero o negativos. En algunos casos, el estado de oxidación promedio de un elemento es una fracción, como 8/3 para hierro en magnetita Fe3O4. Se informa que el estado de oxidación más alto conocido es +9 en el catión tetroxoiridio (IX) (IrO4+). Se predice que incluso un estado de oxidación de +12 puede ser alcanzado por el uranio en el inusual hexóxido UO6. El estado de oxidación más bajo es -5, como para el boro en Al3BC.
En la nomenclatura inorgánica de Stock, el estado de oxidación se representa con un número romano colocado después del nombre del elemento dentro del paréntesis o como un superíndice después del símbolo del elemento, p. Óxido de hierro (III).
El término oxidación fue utilizado por primera vez por Antoine Lavoisier para referirse a la reacción de una sustancia con oxígeno. Mucho más tarde se comprendió que la sustancia, al oxidarse, pierde electrones, y se amplió el significado para incluir otras reacciones en las que se pierden electrones, independientemente de que haya oxígeno involucrado. El aumento del estado de oxidación de un átomo, a través de una reacción química, se conoce como oxidación; una disminución en el estado de oxidación se conoce como una reducción. Tales reacciones implican la transferencia formal de electrones: una ganancia neta de electrones es una reducción y una pérdida neta de electrones es una oxidación. Para elementos puros ya sean monoatómicos, poliatómicos o metálicos, el estado de oxidación es cero.
12.2 Carga formal
Al asignar cargas formales a los átomos en HCl, ignoramos la electronegatividad; los electrones en los enlaces se asignan por igual a los dos átomos enlazados. En este caso, el Cl tiene siete electrones asignados, al igual que el átomo neutro de Cl, y el H tiene un electrón asignado. Por lo tanto, las cargas formales tanto de Cl como de H en este compuesto son 0. Sin embargo, eso tampoco puede ser verdadero, nuevamente por los efectos de la electronegatividad y las propiedades que la generan como la afinidad electrónica a los núcleos, aunque el cloro no es lo suficientemente fuerte como para atraer permanentemente los electrones del hidrógeno sobre sí mismo, sí es lo suficientemente poderoso como para atraerlos la mayoría del tiempo. En otras palabras, la carga formal y el estado de oxidación son dos visiones extremas de una situación que es intermedia entre ambas.
12.3 Carga real
Ni el número de oxidación ni la carga formal dan una descripción precisa de las cargas reales de los átomos porque los números de oxidación exageran el papel de la electronegatividad y las cargas formales lo ignoran. Parece razonable que los electrones en los enlaces covalentes se distribuyan de acuerdo con las electronegatividades relativas de los átomos enlazados. La electronegatividad reportada en la mayoría de las tablas periódicas de mano para el cloro Cl es de 3.0, mientras que el valor de electronegatividad de H es 2.1. Por lo tanto, se podría esperar que el átomo de Cl más electronegativo tenga aproximadamente 3.0/(3.0 + 2.1) = 0.59 de la carga eléctrica en el par enlazante, mientras que el átomo de H tendría 2.1/(3.0 + 2.1) = 0.41 de la carga.
Debido a que el enlace consta de dos electrones, la participación del átomo de Cl es 0.59 x 2e = 1.18e, o 0.18e más que el átomo de Cl neutro. Esto da lugar a una carga negativa parcial de 0.18- en Cl y, por tanto, a una carga positiva parcial de 0.18+ en H. (Observe de nuevo que colocamos los signos más y menos antes de la magnitud al escribir los números de oxidación y las cargas formales, pero después de la magnitud por escrito los cargos reales.)
12.4 Momento dipolar
El momento dipolar del HCl da una medida experimental de la carga parcial de cada átomo. El momento dipolar de HCl corresponde a una carga parcial de 0.178+ en H y 0.178- en Cl, lo que está muy de acuerdo con nuestra aproximación simple basada en electronegatividades. Aunque nuestro método de aproximación proporciona números aproximados para la magnitud de la carga de los átomos, la relación entre las electronegatividades y la separación de carga es generalmente más complicada. Se han desarrollado algoritmos informáticos que emplean los principios de la mecánica cuántica para obtener estimaciones más precisas de las cargas parciales de los átomos, incluso en moléculas complejas.
13. Resonancia
|| REGRESAR AL INDICE ||
A veces nos encontramos con moléculas e iones en los que la disposición de los átomos determinada experimentalmente no está adecuadamente descrita por una única estructura de Lewis dominante.
Ejemplo.Determinar las posibles estructuras de Lewis y su representación de resonancia.
La solución por lo tanto es:

Figura 13.1. Representaciones alternativas de una estructura de resonancia. Si bien la química La Ciencia Central aconseja la forma entre corchetes, la verdad es que la forma con enlace segmentado es mas popular y nos ayuda a ver que la superposición de enlaces afectará la distancia entre átomos.
Si bien se nos dice que cada estructura de Lewis alternativa tiene su propia identidad, al igual que el color azul y el color amarillo deben mantener su propia identidad cuando crean el fantasmagórico color verde, que sólo una ilusión de la combinación de los colores anteriores, la verdad es que la superposición de los enlaces en la estructura resonante nos ayuda a ver qué, la distancia entre los núcleos de los enlaces en una resonancia es intermedia a la que sería, si los dos núcleos estuvieran enlazados por un enlace simple, o por un enlace doble verdadero. Los enlaces dobles atraen más a los núcleos, los enlaces de resonancia están en el medio, y el enlace simple es el que mantiene a los núcleos más alejados.
La disposición real de los electrones en moléculas como el O3 debe considerarse como una combinación de dos (o más) estructuras de Lewis, una superposición cuántica en la que los dos estados de una misma cosa se manifiestan de manera simultánea en el mismo espacio-tiempo. Por analogía con la pintura verde, la molécula tiene su propia identidad separada de las estructuras de resonancia individuales. Por ejemplo, la molécula de ozono siempre tiene dos enlaces O - O equivalentes cuyas longitudes son intermedias entre las longitudes de un enlace simple oxígeno-oxígeno y un enlace doble oxígeno-oxígeno. Otra forma de verlo es decir que las reglas para dibujar estructuras de Lewis no nos permiten tener una única estructura dominante para la molécula de ozono. Por ejemplo, no hay reglas para dibujar medios enlaces. Podemos eludir esta limitación dibujando dos estructuras de Lewis equivalentes que, cuando se promedian, suman algo muy parecido a lo que se observa experimentalmente.
Los enlaces resonantes son más comunes de lo que usted podría esperar, por ejemplo, la gran mayoría de los aniones entre oxígeno y no metales (incluye algunos metales de transición como el manganeso o el cromo en sus estados de oxidación más elevados) generan estructuras de resonancia. esta resonancia hace que estos iones negativos sean extraordinariamente estables, y se comporten como una sola entidad en muchas reacciones químicas como las de desplazamiento y doble desplazamiento.
Ejemplo.Determinar las estructuras de resonancia el anión NO3-
Se sabe que la estructura de resonancia debe ser un estado superpuesto porque la distancia de enlace de N a cualquier oxígeno es la misma. Por lo anterior podemos concluir que, para identificar una estructura de resonancia, debe ser posible identificar la generación de estructuras de Lewis completamente alternativas y completamente simétricas. Hay algunas moléculas o iones para los que todas las estructuras de Lewis posibles pueden no ser equivalentes; en otras palabras, una o más estructuras de resonancia son más dominantes que otras. Encontraremos ejemplos de esto a medida que avancemos.
Química la Ciencia Central 13
Muestra 8.10. ¿Cuál se prevé que tenga los enlaces azufre-oxígeno más cortos, SO3 o SO32- ?
Práctica 8.10.2. Dibuje dos estructuras de resonancia equivalentes para el ion formiato, HCO2-.
13.1 Resonancia del benceno
La resonancia es un concepto importante para describir el enlace en moléculas orgánicas, particularmente en moléculas orgánicas aromáticas, una categoría que incluye el hidrocarburo benceno, C6H6. Los seis átomos de C están unidos en un anillo hexagonal y un átomo de H está unido a cada átomo de C. Podemos escribir dos estructuras de Lewis dominantes equivalentes para el benceno, cada una de las cuales satisface la regla del octeto. Estas dos estructuras están en resonancia:
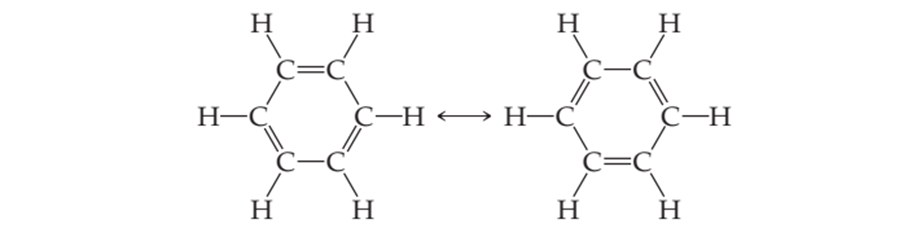
Figura 13.2. Isómeros ideales del benceno.
Tenga en cuenta que los dobles enlaces están en diferentes lugares en las dos estructuras. Cada una de estas estructuras de resonancia muestra tres enlaces simples carbono-carbono y tres enlaces dobles carbono-carbono. Sin embargo, los datos experimentales muestran que los seis enlaces C - C tienen la misma longitud, 1.40 Å, intermedia entre las longitudes de enlace típicas para un enlace simple C - C (1.54 Å) y un enlace doble C = C (1.34 Å). Cada uno de los enlaces C - C del benceno se puede considerar como una combinación de un enlace simple y un enlace doble. El benceno se representa comúnmente omitiendo los átomos de hidrógeno y mostrando solo el marco de carbono-carbono con los vértices sin etiquetar. En esta convención, la resonancia en la molécula se representa por dos estructuras separadas por una flecha de dos puntas o por una notación abreviada en la que dibujamos un hexágono con un círculo dentro:

Figura 13.3. Notación abreviada de la resonancia del benceno.
La notación abreviada nos recuerda que el benceno es una mezcla de dos estructuras de resonancia; enfatiza que los dobles enlaces C = C no pueden asignarse a bordes específicos del hexágono. Los químicos usan ambas representaciones de benceno indistintamente. La disposición de los enlaces en el benceno confiere una estabilidad especial a la molécula. Como resultado, millones de compuestos orgánicos contienen el anillo de seis miembros característico del benceno. Muchos de estos compuestos son importantes en bioquímica, en productos farmacéuticos y en la producción de materiales modernos.
14. Desviaciones del octeto
|| REGRESAR AL INDICE ||
La regla del octeto es tan simple y útil en la introducción de los conceptos básicos de vinculación que podría suponer que siempre se obedece. Sin embargo, ya hemos notado que no puede obedecerse para elementos cuyo gas noble más cercano no tiene 8 electrones, o para elementos que no son representativos, como los metales de transición y las tierras raras.
La regla también falla en muchas situaciones que involucran enlaces covalentes. Estas excepciones a la regla del octeto son de tres tipos principales:
👉 Moléculas e iones poliatómicos que contienen un número impar de electrones
👉 Moléculas e iones poliatómicos en los que un átomo tiene menos de un octeto de electrones de valencia
👉 Moléculas e iones poliatómicos en los que un átomo tiene más de un octeto de electrones de valencia.
14.1 Número de electrones extraños
En la gran mayoría de las moléculas y los iones poliatómicos, el número total de electrones de valencia es par y se produce un apareamiento completo de electrones. Sin embargo, en algunas moléculas e iones poliatómicos, como ClO2, NO, NO2 y O2-, el número de electrones de valencia es impar. El emparejamiento completo de estos electrones es imposible y no se puede lograr un octeto alrededor de cada átomo. Por ejemplo, el NO contiene 5 + 6 = 11 electrones de valencia. Las dos estructuras de Lewis más importantes para esta molécula son:

Figura 14.1. Estructura de Lewis para el monóxido de nitrógeno.
14.2 Estado de oxidación y poder de enlace
A parte del caso anterior, los siguientes casos los podremos racionalizar empleando la nociòn de estado de oxidación-poder de enlace. La idea es muy sencilla, el número de enlaces del átomo central será igual al estado de oxidación del átomo correspondiente. A su vez el estado de oxidación lo podremos calcular como la carga relativa del átomo empleando la ley de la conservación de la carga.
14.3 Un octeto inalcanzable
Un segundo tipo de excepción ocurre cuando hay menos de ocho electrones de valencia alrededor de un átomo en una molécula o ion poliatómico. Esta situación también es relativamente rara (con la excepción del hidrógeno y el helio, como ya hemos comentado), y se encuentra con mayor frecuencia en compuestos de boro y berilio.
Por lo general, representamos BF3 únicamente por la estructura de resonancia dominante, en la que solo hay seis electrones de valencia alrededor del boro. El comportamiento químico de BF3 es consistente con esta representación, lo cual implica que el cálculo de cargas formales es una fuerte indicación del nivel de estabilidad o importancia de una estructura de Lewis determinada.
14.4 Mas de ocho electrones de valencia
La tercera y más grande clase de excepciones consiste en moléculas o iones poliatómicos en los que hay más de ocho electrones en la capa de valencia de un átomo. Cuando dibujamos la estructura de Lewis para PF5, por ejemplo, nos vemos obligados a colocar diez electrones alrededor del átomo de fósforo central:
Ejemplo.Dibujar la estructura de Lewis para PF5
Las moléculas y los iones con más de un octeto de electrones alrededor del átomo central a menudo se denominan hipervalentes. Las moléculas hipervalentes se forman solo para los átomos centrales del período 3 e inferior en la tabla periódica. La razón principal de su formación es el tamaño relativamente mayor del átomo central. Por ejemplo, un átomo de P es lo suficientemente grande como para que se le puedan unir cinco átomos de F (o incluso cinco de Cl) sin estar demasiado abarrotado. Por el contrario, un átomo de N es demasiado pequeño para acomodar cinco átomos unidos a él. Debido a que el tamaño es un factor, las moléculas hipervalentes ocurren con mayor frecuencia cuando el átomo central está unido a los átomos más pequeños y electronegativos: F, Cl y O.
La noción de que una capa de valencia puede contener más de ocho electrones también es consistente con la presencia de orbitales d vacíos en átomos del período 3 e inferiores. En comparación, en los elementos del segundo período, solo los orbitales de valencia 2s y 2p están disponibles para el enlace. Sin embargo, el trabajo teórico sobre el enlace en moléculas como PF5 y SF6 sugiere que la presencia de orbitales 3d vacíos en P y S tiene un impacto relativamente menor en la formación de moléculas hipervalentes. La mayoría de los químicos ahora creen que el mayor tamaño de los átomos de los períodos 3 a 6 es más importante para explicar la hipervalencia que la presencia de orbitales d vacíos.
14.5 Hipervalencia opcional
Por último, hay estructuras de Lewis en las que puede que tenga que elegir entre satisfacer la regla del octeto y obtener las cargas formales más favorables utilizando más de un octeto de electrones.
Ejemplo.Dibujar la estructura de Lewis sin octeto expandido para H2SO4.
Ejemplo.Dibujar la estructura de Lewis con octeto expandido para H2SO4.
Ejemplo. Dibujar la estructura de Lewis con octeto expandido para H3PO4.
Ejemplo. Dibujar la estructura de Lewis sin octeto expandido para H3PO4.
Ejemplo. Dibujar la estructura de Lewis con octeto expandido para Na2SO4.
En la mayoría de los casos, descargas formales más favorables se obtienen empleando la técnica de el estado de oxidación igual a poder de enlace, aunque tengamos átomos centrales con octetos expandidos. La pregunta es ¿cuál de las dos estructuras es más probable o más importante? Cálculos teóricos recientes basados en la mecánica cuántica sugieren a algunos investigadores que la estructura sin octeto expandido es más importante. Otros investigadores afirman que las longitudes de enlace en el ion son más consistentes con la estructura de octeto expandido como dominante. Este desacuerdo es un recordatorio conveniente de que, en general, múltiples estructuras de Lewis pueden contribuir a la distribución real de electrones en un átomo o molécula, y en muchas ocasiones no es necesario conocer la estructura real de una molécula para poder realizar otro tipo de cálculos químicos.
14.6 Valencia diferencial
Hasta ahora hemos visto ejemplos de estructuras de Lewis donde el poder de enlace de todos los átomos del mismo elemento es el mismo, yo sin embargo esto no es necesariamente cierto. TT un ejemplo bastante importante son los no metales del grupo 17 en sus estados de oxidación 3 y 5.
Ejemplo. Dibujar la estructura de Lewis del Cl2O3
Ejemplo. Dibujar la estructura de Lewis del Cl2O5
Los ejemplos anteriores nos demuestran que, yo los 2 átomos de cloro tendrán un poder de enlace diferencial en la estructura de Lewis, y que el poder de enlace no necesariamente es el mismo valor del estado de oxidación. De hecho, también se hace evidente que el oxígeno no siempre necesita entablar dos enlaces. Hola esos presupuestos erróneos pueden a llevarnos a plantear fórmulas estructurales de Lewis que son incorrectas, o aunque sean correctas experimentalmente están mal justificadas.
15. Enlace covalente coordinado
|| REGRESAR AL INDICE ||
El enlace covalente coordinado es el que tiene lugar cuando el par de electrones a compartir los suministra un solo de los átomos involucrados. Este enlace es semi polar, por cuyo motivo se lo llamó a veces dativo, calificativo inapropiado, ya que en realidad los electrones no son dados, sino compartidos en forma desigual.
El enlace covalente coordinado ocurrirá generalmente cuando un átomo posee un par de electrones no en las antes pero que son altamente electronegativos, por lo que aprenderán a capturar partículas positivas, especialmente protones ácidos que provengan ya sea de un ácido o de la autodisociación del agua, lo cual generará guión de carga positiva.
el grupo de compuestos que está sometido a el enlace covalente coordinado más importante son aquellos que poseen nitrógenos con electrones no enlazados, de los cuales la sustancia tipo con la cual generamos los ejemplos más comunes es el amoniaco:

Figura 15.1. Estructura de Lewis del enlace coordinado del grupo amonio.
Cuando se generó una reacción con un ácido se puede generar una reacción a 2 pasos, en el primer paso el amoniaco absorbe el protón para transformarse en el ion amonio, y en el segundo paso absorbe el año negativo original del ácido para convertirse en un compuesto de coordinación iónico:

Esa reacción puede ser particularmente útil ya que muchos ácidos manifiestan propiedades covalentes como la volatilidad a presión y temperatura constante, por lo que convertirlos a un compuesto de coordinación que exhibe propiedades iónicas evita que éstos se volatilicen en condiciones normales de presión y temperatura, lo cual los hace más fáciles de almacenar, y transportar. Eso es particularmente conveniente ya que muchos alcaloides (compuestos orgánicos que contienen nitrógenos que se comportan como amoniaco), que son los compuestos activos de muchos medicamentos, son altamente volátiles y por lo tanto no se podrían almacenar durante largos periodos de tiempo si no se pudieran convertir a compuestos de coordinación con propiedades iónicas.
16. Enlace metálico
|| REGRESAR AL INDICE ||
A principios de la década de 1900, Paul Drüde ideó la teoría del enlace metálico del "mar de electrones" al modelar los metales como una mezcla de núcleos atómicos (núcleos atómicos = núcleos positivos + capa interna de electrones) y electrones de valencia. Los enlaces metálicos ocurren entre los átomos de metales. Mientras que los enlaces iónicos unen metales con no metales, y los covalentes a no-metales consigo mismos, los enlaces metálicos unen la mayor parte de los átomos metálicos. Una hoja de papel de aluminio y un alambre de cobre son lugares donde se puede ver la unión metálica en acción.
Los metales tienden a tener puntos de fusión y puntos de ebullición altos, lo que no es compatible con el enlace covalente, lo que sugiere fuertes enlaces entre los átomos. Incluso un metal blando como el sodio (punto de fusión 97.8 °C) se funde a una temperatura considerablemente más alta que el elemento (neón) que le precede en la tabla periódica. Cuando los átomos de sodio se juntan, el electrón en el orbital atómico 3s de un átomo de sodio comparte espacio con el electrón correspondiente en un átomo vecino para formar un orbital molecular, de la misma manera que se forma un enlace covalente.
La diferencia, sin embargo, es que cada átomo de sodio está siendo tocado por otros ocho átomos de sodio, y el intercambio ocurre entre el átomo central y los orbitales 3s en todos los otros ocho átomos. Cada uno de estos ocho está siendo tocado a su vez por ocho átomos de sodio, que a su vez son tocados por ocho átomos, y así sucesivamente, hasta que haya absorbido todos los átomos en ese trozo de sodio. Todos los orbitales 3s en todos los átomos se superponen para dar una gran cantidad de orbitales moleculares que se extienden por toda la pieza de metal. Tiene que haber una gran cantidad de orbitales moleculares, por supuesto, porque cualquier orbital solo puede contener dos electrones.
Los electrones pueden moverse libremente dentro de estos orbitales moleculares metálicos, por lo que cada electrón se separa de su átomo original. Se dice que los electrones están deslocalizados. El metal se mantiene unido por las fuertes fuerzas de atracción entre los núcleos positivos y los electrones deslocalizados.

Figura 16.1. Enlace metálico: el modelo del mar de electrones: núcleos atómicos positivos (círculos naranjas) rodeados por un mar de electrones deslocalizados (círculos amarillos).
Esto a veces se describe como "una matriz de iones positivos en un mar de electrones". Si vas a usar esta analogía, ¡cuidado! ¿Un metal está formado por átomos o iones? Está hecho de átomos. Cada centro positivo en el diagrama representa todo el resto del átomo, excepto el electrón externo, pero ese electrón no se ha perdido; puede que ya no tenga un vínculo con un átomo en particular, pero todavía está allí en la estructura. Por lo tanto, el sodio metálico se escribe como Na, no como Na+. Adicionalmente cuando un átomo de sodio pierde un electrón éste es incapaz de mantenerse unido a la matriz metálica y por lo tanto es expulsado inmediatamente, lo cual explica porque los procesos de oxidación implican la pérdida de masa de un metal.
En un metal fundido, el enlace metálico todavía está presente, aunque la estructura ordenada se haya roto. El enlace metálico no se rompe por completo hasta que el metal hierve. Eso significa que el punto de ebullición es en realidad una mejor guía para la fuerza del enlace metálico que el punto de fusión. Al fundirse, la unión se afloja, no se rompe. La fuerza de un enlace metálico depende de tres cosas:
👉 El número de electrones que se deslocalizan del metal.
👉 La carga del catión (metal).
👉 El tamaño del catión.
Un enlace metálico fuerte será el resultado de más electrones deslocalizados, lo que hace que aumente la carga nuclear efectiva de los electrones en el catión, lo que hace que el tamaño del catión sea más pequeño. Los enlaces metálicos son fuertes y requieren una gran cantidad de energía para romperse y, por lo tanto, los metales tienen puntos de fusión y ebullición altos. Una teoría del enlace metálico debe explicar cómo puede ocurrir tanto enlace con tan pocos electrones (ya que los metales están ubicados en el lado izquierdo de la tabla periódica y no tienen muchos electrones en sus capas de valencia). La teoría también debe tener en cuenta todas las propiedades químicas y físicas únicas de un metal.
16.1 Triángulo de van Arkel-Ketelaar
Anteriormente, argumentamos que los enlaces entre átomos pueden clasificarse como un rango de enlaces posibles entre enlaces iónicos (transferencia de carga total) y enlaces covalentes (electrones compartidos por completo). Cuando dos átomos de electronegatividades ligeramente diferentes se juntan para formar un enlace covalente, un átomo atrae los electrones más que el otro; esto se llama enlace covalente polar. Sin embargo, los enlaces "iónicos" y "covalentes" simples son conceptos idealizados y la mayoría de los enlaces existen en un continuo bidimensional descrito por el Triángulo de van Arkel-Ketelaar.

Figura 16.2. El triángulo de van Arkel-Ketelaar traza la diferencia en electronegatividad ( Δχ ) y la electronegatividad promedio en un enlace ( ∑χ ). La región superior es donde los enlaces son principalmente iónicos, la región inferior izquierda es donde el enlace es metálico y la región inferior derecha es donde el enlace es covalente.
Los triángulos de enlace o triángulos de van Arkel-Ketelaar (llamados así por Anton Eduard van Arkel y J. A. A. Ketelaar) son triángulos que se utilizan para mostrar diferentes compuestos en diversos grados de enlace iónico, metálico y covalente. En 1941, van Arkel reconoció tres materiales extremos y tipos de unión asociados. Usando 36 elementos del grupo principal, como metales, metaloides y no metales, colocó enlaces iónicos, metálicos y covalentes en las esquinas de un triángulo equilátero, así como especies intermedias sugeridas. El triángulo de enlace muestra que los enlaces químicos no son solo enlaces particulares de un tipo específico. Más bien, los tipos de enlaces están interconectados y diferentes compuestos tienen diferentes grados de diferente carácter de enlace (por ejemplo, enlaces covalentes polares).
16.2 Propiedades metálicas
Los metales tienen varias cualidades que son únicas, como la capacidad de conducir electricidad y calor, una energía de ionización baja y una electronegatividad baja (por lo que ceden electrones fácilmente para formar cationes). Sus propiedades físicas incluyen una apariencia lustrosa (brillante), y son maleables y dúctiles. Los metales tienen una estructura cristalina pero pueden deformarse fácilmente, es decir, pueden doblarse. En este modelo, los electrones de valencia son libres, deslocalizados, móviles y no están asociados con ningún átomo en particular. Este modelo puede dar cuenta de:
👉 Conductividad
Dado que los electrones son libres, si los electrones de una fuente externa fueran empujados hacia un extremo de un cable de metal, los electrones se moverían a través del cable y saldrían por el otro extremo a la misma velocidad (la conductividad es el movimiento de carga).
👉 Maleabilidad y ductilidad:
El modelo del mar de electrones de los metales no solo explica sus propiedades eléctricas, sino también su maleabilidad y ductilidad. El mar de electrones que rodea a los protones actúa como un colchón, por lo que cuando se martilla el metal, por ejemplo, la composición general de la estructura del metal no se daña ni cambia. Los protones pueden reorganizarse, pero el mar de electrones se ajustará a la nueva formación de protones y mantendrá intacto el metal. Cuando una capa de iones en un mar de electrones se mueve a lo largo de un espacio con respecto a la capa debajo de ella, la estructura cristalina no se fractura, sino que solo se deforma.
👉 Capacidad calorífica:
Esto se explica por la capacidad de los electrones libres para moverse por el sólido.
👉 Brillo
Los electrones libres pueden absorber fotones en el "mar", por lo que los metales tienen un aspecto opaco. Los electrones en la superficie pueden hacer rebotar la luz a la misma frecuencia que la luz golpea la superficie, por lo tanto, el metal parece estar brillante.
Sin embargo, estas observaciones son solo cualitativas y no cuantitativas, por lo que no se pueden probar. La teoría del "Mar de electrones" se presenta hoy solo como un modelo simplificado de cómo funciona el enlace metálico.
16.3 Efecto de los electrones de valencia en las propiedades metálicas
Se sabe que el punto de fusión del magnesio (650 °C) es más alto que el del sodio (97.79 °C). El magnesio tiene la estructura electrónica externa 3s2. Ambos electrones se deslocalizan, por lo que el "mar" tiene el doble de densidad de electrones que el sodio. Los "iones" restantes también tienen el doble de carga (si va a usar esta vista particular del enlace metálico) y, por lo tanto, habrá más atracción entre los "iones" y el "mar".
De manera más realista, cada átomo de magnesio tiene 12 protones en el núcleo en comparación con los 11 del sodio. En ambos casos, el núcleo está protegido de los electrones deslocalizados por la misma cantidad de electrones internos: los 10 electrones en los orbitales 1s2 2s2 2p6. Eso significa que habrá una atracción neta del núcleo de magnesio de 2+, pero solo 1+ del núcleo de sodio.
Así que no sólo habrá un mayor número de electrones deslocalizados en el magnesio, sino que también habrá una mayor atracción hacia ellos desde los núcleos de magnesio. Los átomos de magnesio también tienen un radio ligeramente más pequeño que los átomos de sodio, por lo que los electrones deslocalizados están más cerca de los núcleos. Cada átomo de magnesio también tiene doce vecinos cercanos en lugar de los ocho del sodio. Ambos factores aumentan aún más la fuerza del vínculo.
Los metales de transición tienden a tener puntos de fusión y puntos de ebullición particularmente altos. La razón es que pueden involucrar a los electrones 3d en la deslocalización así como a los 4s. Cuantos más electrones pueda involucrar, más fuertes tienden a ser las atracciones y más altos los puntos de fusión.
17. Teoría del enlace de valencia
|| REGRESAR AL INDICE ||
Una vez resuelto el asunto de la distribución de los electrones en átomos individuales, aún quedaba el asunto del enlace químico ¿por qué el enlace químico existe? ¿porque los átomos individuales se unen y no permanecen separados como en los gases nobles? o ¿cómo definir la geometría de la molécula?

Figura 17.1. Energía de la molécula de hidrógeno en términos de la distancia entre hidrógenos. La energía con respecto a la distancia de dos átomos de hidrógeno establece un punto de equilibrio en E2 donde la energía de la molécula como un todo es menor a la de los dos átomos por separado o demasiado unidos, de esta forma las moléculas se forman debido a la generación de distancias donde la sustancia existe como una entidad de baja energía muy estable.
Adicionalmente no se explica porque enlaces de un mismo tipo puedan tener propiedades diferentes, por ejemplo, los enlaces que unen la molécula de gas de hidrógeno H-H y la molécula del gas de flúor F-F son en esencia un enlace covalente perfecto de tipo sigma, pero aun cuando son el mismo tipo de enlace, propiedades físicas como la energía de enlace y la distancia de enlace son diferentes. Estos y muchos otros fenómenos no pueden ser explicados por la teoría de Lewis y sus derivados que vimos en el enlace químico clásico.
Para un entendimiento más completo del enlace químico es necesario mirar la teoría cuántica, aunque sea en términos cualitativos. Hasta el momento se han formulado dos teorías mecánico cuánticas para describir el enlace covalente. La primera es la teoría del enlace de valencia que asume que los electrones en una molécula ocupan orbitales atómicos de los átomos individuales. Esto nos permite mantener una imagen de los átomos individuales durante la formación de la molécula, o en otras palabras, predecir geometrías empleando solo el átomo central como guía.
La segunda teoría se denomina orbital molecular, la cual asume la formación de orbitales moleculares a partir de los orbitales atómicos. Ninguna de las dos teorías es perfecta o explica todos los aspectos del enlace, pero tomadas en conjunto han permitido el entendimiento de las propiedades medibles de muchas moléculas. Comencemos la discusión de la teoría del enlace de valencia discutiendo la formación de la molécula de hidrógeno gaseoso H-H. La teoría de Lewis describe la formación de este enlace en términos del emparejamiento de electrones provenientes de cada uno de los átomos de hidrógeno.
Ahora ingresa la parte cuántica. Individualmente cada hidrogeno debe poseer un solo electrón en el nivel 1s, el cual tiene una forma de probabilidad esférica. El electrón en dicha posición mantiene un estado de energía determinado que podríamos llamar como estado inicial o E1. Debido a que es el estado de inicio arbitrariamente definimos que E1 es igual a cero y a partir de él se miden cambios de energía. El enlace sigma entre orbitales 1s se da por el sobrelapamiento del volumen probable en el cual se puede encontrar un electrón, creando un volumen efectivo en el cual es muy probable encontrar los electrones de los átomos independientes. Una vez en medio, la carga electrostática de los dos electrones mantiene unido a los núcleos positivos de ambos átomos.
La pregunta es qué pasa con la energía cuando los orbitales 1s se sobrelapan. La respuesta experimental es una emisión energética, en otras palabras, el nuevo orbital de la molécula es mucho más estable, o lo que es lo mismo, porta menos energía que los orbitales 1s por separado. En el punto más estable los núcleos se encuentran bastante cerca unidos por los dos electrones, punto de denominamos E2. Ahora, más allá de E2 los núcleos no se acercan más ya que a la distancia mínima de enlace las cargas de los protones empiezan a interactuar entre sí en los dos átomos independientes, en otras palabras, los dos núcleos positivos empiezan a repelerse, por lo que para acercar más los núcleos hay que adicionar energía, y para que se toquen la energía sigue una asíntota ascendente que tiende al infinito, pero que no es infinito. De hecho, tanto en las bombas de hidrogeno como en los núcleos de las estrellas dicha energía es alcanzando y los núcleos del hidrogeno se tocan.
Ahora, la repulsión provocada por los núcleos explicaría parcialmente la diferencia de propiedades del enlace tipo sigma perfecto. En la molécula de flúor la distancia mínima antes de que los núcleos empiecen a repelerse entre sí es mayor debido a que el núcleo del flúor posee una mayor carga electrostática positiva, por lo que se repelerá con más fuerza. En ese sentido, la teoría del enlace de valencia es una teoría para el estado energético del enlace con respecto a las cargas electrostáticas, no solo de los electrones, sino también de los núcleos, lo cual da una imagen más clara de la formación del enlace químico. En términos más generales, la teoría del enlace de valencia establece que los enlaces se forman entre átomos cuyas moléculas alcanzan un estado energético menor al de los átomos por separado.
17.1 Enlaces covalentes por hibridación.
Los enlaces covalentes pueden diferenciarse en dos categorías básicas, el enlace sigma y el enlace pi. Reconocer las diferencias entre estos dos tipos de enlace es crucial para poder entender la geometría molecular, ya que uno de los enlaces influye fuertemente en la geometría molecular mientras que el otro no.
17.2 El enlace sigma
En química el enlace covalente tipo sigma es el enlace más fuerte. Estos se forman mediante el sobrelapamiento de los orbitales atómicos para formar orbitales moleculares más estables y de menor energía. Típicamente los enlaces sigma son los primeros en formarse por lo que corresponderán a casi cualquier enlace simple; mientras que los enlaces múltiples contendrán un enlace sigma y varios enlaces pi que son más inestables. Los enlaces sigma pueden rotar subre su propio eje, permitiendo que los átomos unidos puedan rotar como un tornillo en una tuerca.
17.3 El enlace pi
En química los enlaces pi son enlaces covalentes que se forman mediante el sobrelapamiento de dos lóbulos de probabilidad de orbitales que posean como mínimo dos lóbulos, como los orbitales p o los orbitales d. La forma de la onda del orbital molecular es semejante a un anillo que atraviesa a ambos núcleos pero que se desaparece a medida que se acerca a los núcleos.
La letra griega pi hace referencia a los orbitales p debido a que la simetría del orbital pi es la misma que la del orbital p cuando se los ve desde el plano del enlace. Los orbitales p son los típicos orbitales que forman los enlaces pi aunque también pueden hacerlo los orbitales d.
Los orbitales tipo pi son usualmente más débiles e inestables que los orbitales sigma debido a que portan una mayor cantidad de energía. Adicionalmente debido a su forma de anillo, los enlaces pi no pueden rotar libremente lo que torna a las moléculas unidas por ellos fijas, pero tendientes al rompimiento, esto implica una mayor reactividad. En cuanto a la geometría molecular el enlace pi va a seguir la dirección del primer enlace sigma, por lo que los electrones contenidos en dicho enlace no afectaran de forma notable a los electrones de otros enlaces o a los orbitales no enlazantes.
17.4 Teoría de repulsión de pares de electrones de valencia TRePEV
Nuevamente las cuestiones con el enlace químico son muy complicadas por lo que solo nos enfocamos a algunos métodos heurísticos que permiten entender el modo en que se organizan las moléculas. Las moléculas unidas por enlaces covalentes pueden ser explicadas bastante bien mediante la fusión de dos modelos que aunque no están directamente relacionados, tradicionalmente se enseñan juntos: la Hibridación Molecular “HM” y la Teoría de Repulsión de Pares de Electrones de Valencia “TRePEV”.
El proceso en realidad es simple, (1) hay que determinar la configuración electrónica con los cuatro números cuánticos para el ultimo nivel de energía del elemento central de una molécula, (2) realizar la hibridación de forma tal que se creen los orbitales necesarios para el número de enlaces requerido para expl9icar la molécula, (3) determinar la cantidad de enlaces y pares de electrones no enlazantes, y finalmente (4) determinar la geometría molecular más probable.

Figura 17.2. Geometrías moleculares. En la figura tenemos los átomos centrales en rojo, que son aquellos de quienes modelamos su distribución electrónica, en verde los átomos periféricos y en azul los pares de electrones no enlazantes. Aunque la cantidad de formas es variada, las que más nos importan son las que tetrahedro, las del trigonal planar y las del lineal, ya que serán relevantes en los capítulos de química orgánica.
Como todos estos modelos químicos hay moléculas que no la cumplen, pero usamos este método debido a que muchas moléculas SI la cumplen. En términos simples TRePEV establece que las moléculas covalentes son entidades tridimensionales cuyos enlaces y orbitales no enlazantes se van a organizar espacialmente de forma tal que cada uno se encuentre lo más alejado del resto como sea físicamente posible.
En ese sentido la geometría se organiza en base a un centro molecular (E) que usualmente es el átomo menos numeroso en la molécula y una serie de posiciones que lo orbitan (X). Estas posiciones pueden ser de dos tipos, enlaces tipo sigma y orbitales no enlazantes. Los orbitales tipo pi no afectan la geometría molecular de forma evidente, por lo que no los tomamos realmente en cuenta a la hora de definir una geometría molecular en especial.
Como pueden haberlo notado, el problema con aplicar la TRePEV es identificar la cantidad de posiciones que rodean al centro molecular, ya que hasta el momento no hemos introducido ninguna técnica que nos permita identificar la presencia y cantidad de orbitales no enlazantes. Para resolver el problema anterior hay que emplear el concepto de hibridación de orbitales que es una teoría independiente, pero que por tradición se enseña junto a la TRePEV ambas como parte de la Teoría del enlace de Valencia.
18. Hibridaciones sp, sp2 y sp3 en la teoría del enlace de valencia
|| REGRESAR AL INDICE ||
18.1 Hibridación de orbitales sp
La hibridación se basa en la idea de que los orbitales individuales se combinan y alinean en un estado de energía homogéneo, una vez alineados se denominan orbitales sp. No todos los cuatro orbitales se deben hibridar al mismo tiempo, ya que algunos elementos no tienen suficientes electrones para hacerlo. Pero para explicarlo mejor hay que hacer ejemplos:
👉 Hidruro de berilio
Vamos a predecir la geometría molecular y las propiedades electrostáticas de la molécula de hidruro de berilio , Primero que todo vamos a dibujar la molécula según las indicaciones que recibimos en los estados de oxidación, el berilio solo puede formar dos enlaces y el hidrogeno solo uno. La organización de esta molécula puede ser lineal o angular. Para conocer cual hipótesis es más aceptable, lo primero que debemos hacer es tener la configuración electrónica del berilio con cuatro números cuánticos: Be=[He]2s(↑↓), Ahora el punto es este, el berilio tiene un orbital saturado en el subnivel s que no podían formar un enlace covalente, mientras que tiene tres orbitales p vacíos [He]2s(↑↓) p(↑) (↓) (↓). Para formar un enlace se necesita un orbital con un solo electrón que reciba el electrón del átomo acompañante o átomos acompañantes, en este caso necesitamos espacio para dos electrones que vienen de dos hidrógenos. Para lograrlo se crean orbitales híbridos igual a la cantidad de enlaces, en este caso dos y se distribuyen los electrones de forma que solo llenen medio orbital [He]2sp(↑↓) (↑↓) p(↑↓) (↑↓).

Figura 18.1. Dos hipótesis para la geometría del BeH2 Hipotesis lineal (izquierda) y angular (derecha) para la geometría de la molécula de hidruro de berilio.
Con los dos orbitales enlazantes sp el berilio puede recibir dos electrones ajenos, lo cual equivale a poder formar dos enlaces tipo sigma BeH2=[He] 2sp(↑↓)(↑↓). Ahora sabemos que hay dos lóbulos de electrones alrededor del berilio ambos terminando en átomos, por lo que buscamos en la (Figura 17.2), cuál será la geometría para un átomo central con dos átomos periféricos sin nada más extra, y la respuesta es lineal. Por lo que la geometría del hidruro de berilio es lineal. Una vez señalada la geometría podemos concluir que la molécula de hidruro de berilio es una molécula plana, y como en sus extremos se encuentra el mismo átomo no hay una polarización, por lo tanto, no generará interacciones moleculares electrostáticas débiles “es decir no se comporta como un pequeño imán”.
👉 Etino
Generalmente la geometría molecular se describe en términos de moléculas muy simples, sin embargo, es importante entender un concepto a cerca de las hibridaciones, y es que no necesariamente son completas. De hecho, cuando existen enlaces múltiples, las hibridaciones pueden ser diferentes. A lo que me refiero, es que un mismo elemento puede hibridar de manera diferente, para dar origen a un diferente arreglo y a una diferente geometría, tal es el caso del carbono, cuyas geometrías variantes son de importancia crucial en
la química orgánica. Y es debido a esa importancia que he decidido colocar la hibridación del carbono sp en esta sección. Bien, en primera instancia debemos escribir la fórmula de la molécula y ubicar el átomo central, pues es el quien determina la geometría de la molécula. La fórmula del Etino es C2H2), luego, determinamos cuantos enlaces debemos explicar. El carbono siempre posee 4 enlaces potenciales, a menos que esté haciendo la molécula de monóxido de carbono. Cada hidrógeno tiene tan solo un enlace. Por lo tanto, cada carbono tendrá tres enlaces entre si y un enlace al hidrógeno. Sin embargo, eso nos genera varias hipótesis de geometría:

Figura 18.2. Hipótesis para la geometría molecular del etino.
Como detalle adicional, cuando tenemos enlaces múltiples solo el primer enlace va a ser hibrido de baja energía, los demás enlaces se forman entre orbitales p o d que no hibridan. En consecuencias la hibridación que debemos lograr debe explicar dos enlaces híbridos estables sp y dos orbitales p, cada uno de los cuales con un solo electrón que pueda formar enlace. Realizaremos la hibridación desde el punto de vista de uno de los carbonos. Primero buscamos la configuración electrónica del carbono C=[He]2s(↑↓)p(↑)(↑)( ), en esta forma el carbono solo puede explicar dos enlaces, por lo que comenzamos con la hibridación. Dado que hay tres enlaces entre carbonos, uno de ellos será sigma con hibridación sp, y los otros serán pi no hibridados, además del enlace al carbono que también es sigma, por lo que requerimos dos orbitales sp, y dos orbitales p semi-llenos, lo cual se logra simplemente moviendo el electrón de orbital s que desaparece C=[He]2sp(↑)(↑)p(↑)(↑). La clave para la geometría molecular es que los orbitales p no cuentan como lóbulos independientes, porque están alineados con uno de los orbitales sp, y por ende solo tenemos dos lóbulos para cada carbono, lo cual implica una geometría lineal.
18.2 Hibridación sp2
En la hibridación sp2 se formarán tres orbitales sp2 híbridos.
👉 Trihidruro de boro
El boro tiene un estado de oxidación de 3 (no nos importa) por lo que va a formar tres enlaces covalentes, mientras que el hidrogeno solo puede formar un enlace covalente. La fórmula molecular de la sustancia es . El boro es el átomo central y por ende de quien nos fijamos B=[He]2s(↑↓)p(↑)( )( ), tal como se nos presenta, el boro solo puede explicar un enlace, pero si un electrón salta del orbital s y quedan tres orbitales sp semi-llenos B=[He]2sp(↑)(↑)(↑)p( ) obtenemos una hibridación de un s y dos p, llamada sp2. En este caso tenemos tres lóbulos cuya geometría en la es la trigonal planar:

Figura 18.3. Geometría trigonal plana.
👉 Eteno
Generalmente la geometría molecular se describe en términos de moléculas muy simples, sin embargo, es importante entender un concepto a cerca de las hibridaciones, y es que no necesariamente son completas. De hecho, cuando existen enlaces múltiples, las hibridaciones pueden ser diferentes. A lo que me refiero, es que un mismo elemento puede hibridar de manera diferente, para dar origen a un diferente arreglo y a una diferente geometría, tal es el caso del carbono, cuyas geometrías variantes son de importancia crucial en la química orgánica. Y es debido a esa importancia que he decidido colocar la hibridación del carbono sp2 en esta sección. Bien, en primera instancia debemos escribir la fórmula de la molécula y ubicar el átomo central, pues es el quien determina la geometría de la molécula ), determinamos cuantos enlaces debemos explicar. El carbono siempre posee 4 enlaces potenciales, a menos que esté haciendo la molécula de monóxido de carbono. Cada hidrógeno tiene tan solo un enlace. Por lo tanto, cada carbono tendrá dos enlaces entre si y un enlace a cada hidrógeno. El problema nuevamente es que podemos pensar en varias geometrías. La clave aquí es que la geometría del primer carbono será igual a la del segundo carbono, pues los hidrógenos serán los átomos periféricos.

Figura 18.4. Geometría trigonal plana de dos centros de geometría, en este caso, dos carbonos.
La configuración del carbono es C=[He]2s(↑↓)p(↑)(↑)( ), en este caso necesitamos explicar tres enlaces sigma con hibridación sp, uno entre carbonos y otro para los hidrógenos, y un enlace pi entre orbitales p entre carbonos C=[He]2sp2(↑)(↑)(↑)p(↑) de esta manera, para los lóbulos solo contamos los sp, ya que el enlace entre orbitales p se alinea con uno de los enlaces sigma, por lo que tenemos tres lóbulos y por ende una geometría trigonal planar en ambos carbonos.
18.3 Hibridación sp3
En este caso todos los orbitales p se convierten en orbitales sp
👉 Metano
La geometría sp3 del carbono es tal vez la más conocida de sus hibridaciones cuando trabaja con el estado de oxidación 4 “generar 4 enlaces”. Esto se debe a que es la geometría más común que adquiere el carbono en los compuestos biológicos de importancia, así como en los hidrocarburos y demás compuestos orgánicos. Bien, en primera instancia debemos escribir la fórmula de la molécula y ubicar el átomo central, pues es el quien determina la geometría de la molécula ).
Al armar la molécula siguiendo los valores de sus estados de oxidación tenemos que el carbono formaría 4 enlaces y cada hidrogeno solo un enlace, lo cual lo induce a uno a dibujar una molécula plana como una cruz, con ángulos de 90° entre los hidrógenos, el problema es que las moléculas viven en un espacio tridimensional y 90° no es la fórmula más alejada cuando hablamos de geometrías NO planas. Miremos cómo funcionaría una hibridación para esta molécula, iniciamos con la configuración del carbono normal C=[He]2s(↑↓)p(↑)(↑)(_), como necesitamos cuatro en laces sigma, debemos hibridar todos los orbitales p normal C=[He]2sp3(↑)(↑)(↑)(↑), con esto obtenemos cuatro lóbulos, al mirar en la (Figura 17.2) lo que obtenemos es un tetraedro, con ángulos entre hidrógenos de aproximadamente 109,5°.

Figura 18.5. Geometría molecular del metano.
👉 El amoníaco
El amoniaco es una molécula interesante, ya que presentará un par de electrones no enlazantes que se organizará en un orbital sigma no enlazante. A pesar de que este orbital no señalará hacia otro átomo, de todas formas, ocupa un lugar en el espacio. Bien, en primera instancia debemos escribir la fórmula de la molécula y ubicar el átomo central, pues es el quien determina la geometría de la molécula (NH3). Determinamos la configuración electrónica del nitrógeno N=[He]2s(↑↓)p(↑)(↑)(↑), ahora debemos explicar tres enlaces sigma a hidrógenos, que requieren orbitales sp, la única forma de generar tres orbitales sp semi-llenos es mediante una hibridación sp3: N=[He]2sp3(↑↓)(↑)(↑)(↑) con lo cual obtenemos cuatro lóbulos, pero uno de ellos no tiene átomo, por lo que la (Figura 17.2) nos indica que estamos tratando con una geometría trigonal piramidal. Esto a demás explica el estado de oxidación normal del nitrógeno como (3).

Figura 18.6. Geometría molecular del amoníaco.
El par de electrones no enlazantes le permite al amoníaco capturar un protón extra como si fuera un anión, por ende, se comporta como una base. En la teoría de Lewis para ácidos y bases, muchas sustancias que quedan con un par de electrones no enlazantes se comportaran de manera semejante y se denominan bases de Lewis.
👉 El agua
Siguiendo los pasos del nitrógeno, el oxígeno también hibrida de modo sp3 cuando requiere realizar dos enlaces a átomos independientes, pero al hacerlo genera orbitales sigma no enlazantes. A diferencia del nitrógeno que solo genera un orbital sigma no enlazante, el oxígeno genera dos orbitales sigma no enlazantes. Bien, en primera instancia debemos escribir la fórmula de la molécula y ubicar el átomo central, pues es el quien determina la geometría de la molécula (H2O). La configuración del oxígeno es O=[He]2s(↑↓)p(↑↓)(↑)(↑), ahora debemos explicar dos enlaces sigma a hidrógenos, que requieren orbitales sp, la única forma de generar dos orbitales sp semi-llenos es mediante una hibridación sp3: O=[He]2sp3(↑↓)(↑↓)(↑)(↑), con lo cual obtenemos cuatro lóbulos, pero dos de ellos no tienen átomos, por lo que la (Figura 17.2) nos indica que estamos tratando con una geometría angular o doblada. Esto a demás explica el estado de oxidación normal del oxígeno como (2).

Figura 18.7. Geometría molecular del agua.
El agua se comporta como una base de Lewis al tener dos pares de electrones no enlazantes, capturando un protón y formando el catión ácido H3O+.
19. Hibridaciones sp3d y sp3d2 en la teoría del enlace de valencia
|| REGRESAR AL INDICE ||
19.1 Hibridación sp3d
Las hibridaciones sp3 son las que permiten explicar los octetos ya que una gran cantidad de moléculas se podrían explicar por ella. En una hibridación sp3 siempre se completan ocho electrones en los orbitales híbridos de valencia, pero como se mencionó antes, no todas las moléculas cumplen octetos. Existen hibridaciones superiores que hacen uso de los orbitales d híbridos para poder ir más allá de los ocho electrones de valencia.
👉 Pentacloruro de fósforo
Bien, en primera instancia debemos escribir la fórmula de la molécula y ubicar el átomo central, pues es el quien determina la geometría de la molécula (PCl5). La configuración electrónica del fósforo es P=[Ne]3s(↑↓)p(↑)(↑)(↑), en una hibridación sp3 solo obtendríamos 3 enlaces, por lo que debemos emplear algunos orbitales d vacíos P=[Ne]3s(↑↓)p(↑)(↑)(↑)d(_)(_)(_)(_)(_), si hibridamos todos los orbitales que podemos con 1 electrón cada uno, obtenemos cinco orbitales híbridos llamados sp3d, P=[Ne]3sp3d(↑)(↑)(↑)(↑)(↑)d(_)(_)(_)(_), e ignoramos los orbitales de que quedan vacíos. En este caso la sustancia queda con cinco lóbulos, todos los cuales tienen un átomo de cloro, por lo que si vemos la (Figura 17.2), la geometría a elegir será trigonal bipiramidal.

Figura 19.1. Geometría trigonal bipiramidal.
👉 Tetrafloruro de azufre
Bien, en primera instancia debemos escribir la fórmula de la molécula y ubicar el átomo central, pues es el quien determina la geometría de la molécula (SF4). Luego escribimos la configuración electrónica del azufre S=[Ne]3s(↑↓)p(↑↓)(↑)(↑), una hibridación sp3 solo me daría dos enlaces y requiero cuatro, así que empleamos un orbital d extra para expandir el poder de enlace S=[Ne]3s(↑↓)p(↑)(↑)(↑)(↑)d(_)(_)(_)(_), de esta manera obtenemos cuatro enlaces y un par de electrones no enlazantes, si analizamos la (Figura 17.2), la geometría a elegir será la de balancín.

Figura 19.2. Geometría de balancín.
👉 Trifluoruro de cloro
Bien, en primera instancia debemos escribir la fórmula de la molécula y ubicar el átomo central, pues es el quien determina la geometría de la molécula (ClF3). Luego escribimos la configuración electrónica del cloro Cl=[Ne]3s(↑↓)p(↑↓)(↑↓)(↑), una hibridación sp3 solo me daría un enlace y requiero tres, así que empleamos un orbital d extra para expandir el poder de enlace Cl=[Ne]3sp3d(↑↓)(↑↓)(↑)(↑)(↑), de esta manera obtenemos tres enlaces y dos pares de electrones no enlazantes, si analizamos la (Figura 17.2), la geometría a elegir será la forma T.

Figura 19.3. Geometría T plana.
19.3 Hibridación sp3d2
Continuamos con las hibridaciones que van más allá de los octetos.
👉 Hexafluoruro de azufre
Bien, en primera instancia debemos escribir la fórmula de la molécula y ubicar el átomo central, pues es el quien determina la geometría de la molécula (SF6). Luego escribimos la configuración electrónica del azufre S=[Ne]3s(↑↓)p(↑↓)(↑)(↑), una hibridación sp3 solo me daría dos enlaces y requiero seis, así que empleamos dos orbitales d extra para expandir el poder de enlace S=[Ne]3sp3d2(↑)(↑)(↑)(↑)(↑)(↑) de esta manera obtenemos seis enlaces sigma, si analizamos la (Figura 17.2), la geometría a elegir será el octaedro.

Figura 19.4. Geometría octaédrica
👉 Pentafluoruro de cloro
Bien, en primera instancia debemos escribir la fórmula de la molécula y ubicar el átomo central, pues es el quien determina la geometría de la molécula (ClF5). Luego escribimos la configuración electrónica del cloro Cl=[Ne]3s(↑↓)p(↑↓)(↑↓)(↑), una hibridación sp3 solo me daría un enlace y requiero cinco, así que empleamos dos orbitales d extra para expandir el poder de enlace Cl=[Ne]33sp3d2 (↑↓)(↑)(↑)(↑)(↑)(↑), de esta manera obtenemos cinco enlaces sigma y un par no enlazante, si analizamos la (Figura 17.2), la geometría a elegir será el cuadrado piramidal.
Figura 19.5. Geometría cuadrado piramidal.
👉 Tetrafluoruro de xenon
Bien, en primera instancia debemos escribir la fórmula de la molécula y ubicar el átomo central, pues es el quien determina la geometría de la molécula (XeF4). Luego escribimos la configuración electrónica del xenón Xe =[Ne]3s(↑↓)p(↑↓)(↑↓)(↑↓), una hibridación sp3 solo me daría ningún enlace y requiero cuatro, así que empleamos dos orbitales d extra para expandir el poder de enlace Xe =[Ne]3sp3d2(↑↓)(↑↓)(↑)(↑)(↑)(↑), de esta manera obtenemos cuatro enlaces sigma y dos pares no enlazantes, si analizamos la (Figura 17.2), la geometría a elegir será la cuadrado planar.

Figura 19.6. Geometría cuadrado planar.
20. Efectos de la geometría molecular
|| REGRESAR AL INDICE ||
20.1 Momento dipolar
El momento dipolar hacen referencia a las cargas parciales que adquieren los miembros de una molécula cuando estos poseen electronegatividades diferentes y/o orbitales con electrones no enlazantes. Aunque existe una fórmula para calcular cuantitativamente el momento dipolar la verdad es que nos es más útil lo cualitativo en los cursos de química general.
Una molécula que posee un momento dipolar se conoce simplemente como una molécula polar, capaz de entablar enlaces más débiles con otras moléculas, lo cual afecta las propiedades físicas del material que componen. Una sustancia sin momento dipolar la conocemos como apolar, es decir sin polos determinados, lo cual impide que forme interacciones moleculares. Cualitativamente podemos definir el momento dipolar como si fueran vectores. En ese sentido para que exista momento dipolar debe haber una suma neta de vectores que no se anule.

Figura 20.1. Análisis de momento dipolar para varias geometrías moleculares. Si las flechas apuntan en direcciones opuestas en un mismo plano “o su suma vectorial da cero” el momento dipolar se cancela y la molécula se comporta como si fuera neutra. Si las flechas apuntan en diferentes ángulos, se generan momentos dipolares, que hacen que se comporte como un pequeño imán.
20.2 Geometrías iónicas
Las moléculas iónicas por su parte no poseen una geometría molecular determinada, aunque si tienen una geometría iónica, y por lo tanto no pueden ser representadas por la teoría del enlace de valencia ni tampoco por la teoría del orbital molecular, ambas teorías están restringidas a los enlaces covalentes. Las moléculas iónicas están compuestas por capas y capas organizadas de acuerdo con sus cargas electrostáticas, lo que hace que todo el sólido sea fuerte, pero a su vez fácil de separar en el agua. En cuanto a los iones poliatómicos emerge el problema de la resonancia e intentar describir la geometría de uno o dos electrones deslocalizados es inútil, es por eso que ellos son descritos en términos de sus fórmulas moleculares simples.
21. Teoría del orbital molecular
|| REGRESAR AL INDICE ||
Aunque la teoría del enlace de valencia que conjuga el modelo de Lewis, las estructuras que construimos con los estados de oxidación, la hibridación de orbitales y la TrePEV es una aproximación importante al entendimiento del enlace químico en términos de la mecánica cuántica no es la única explicación. La simple presunción de que los electrones en una molécula ocupan orbitales atómicos en los átomos individuales puede que sea solo una aproximación, debido a que cada electrón enlazantes en una molécula debe estar en un orbital que sea característico de la molécula como un todo. En algunos casos la teoría del enlace de valencia no puede explicar las propiedades medidas de algunas moléculas. Considere la molécula de oxígeno:

Figura 21.1. Estructura de Lewis para el dioxígeno.
De acuerdo con esta descripción todos los electrones en la molécula de oxígeno están apareados, ya sea en orbitales de enlace o en orbitales no enlazantes, lo cual implicaría que el oxígeno es diamagnético. En electromagnetismo, el diamagnetismo es una propiedad de los materiales que consiste en repeler los campos magnéticos. Es lo opuesto a los materiales paramagnéticos los cuales son atraídos por los campos magnéticos. El fenómeno del diamagnetismo fue descubierto por Sebald Justinus Brugmans que observó en 1778 que el bismuto y el antimonio fueron repelidos por los campos magnéticos (Samuel, n.d.). El término diamagnetismo fue acuñado por Michael Faraday en septiembre de 1845, cuando se dio cuenta de que todos los materiales responden (ya sea en forma diamagnética o paramagnética) a un campo magnético aplicado. Retornando al oxígeno, sin electrones desapareados no debería experimentar atracción por un campo magnético. Sin embargo, las mediciones arrojan que el oxígeno posee en efectos dos electrones desapareados debido a su característico paramagnetismo. Esto sugiere una deficiencia fundamental en la teoría del enlace de valencia, una que justifica la búsqueda de una teoría de enlace que dé cuenta por las propiedades del oxígeno molecular.
El magnetismo y otras propiedades de las moléculas son explicados “a veces” de mejor manera por la segunda teoría del enlace de valencia denominada orbital molecular. La teoría del orbital molecular describe el enlace covalente en términos de orbitales moleculares, que resultan de la interacción de los orbitales atómicos enlazantes y que están asociados a la molécula completa. La diferencia entre un orbital molecular y un orbital atómico es que un orbital atómico está asociado a un solo átomo.
21.2 El equivalente de la regla de Aufbau
En la teoría del orbital molecular los electrones de los átomos serán cedidos a los orbitales de las moléculas, pero la clave es que los orbitales de las moléculas también tienen un orden fijo de llenado:
σ1s()<σ*1s()<σ2s()<σ*2s()<π2py()=π2pz()<π2px()<π*2py()=π*2pz()<π*2px()
Figura 17.2. Energías de enlace de los orbitales moleculares. Estas crecen de izquierda a derecha.
21.4 Orbitales moleculares enlazantes y antienlazantes
Como se pueden dar cuenta, los orbitales moleculares tienen ciertas equivalencias a los orbitales atómicos, por ejemplo, los orbitales s estarán vinculados a los orbitales moleculares (sigma σ), y los orbitales p a los orbitales moleculares (pi π), pero ¿Qué significa el asterisco?
De acuerdo con la teoría del orbital molecular, los orbitales 1s de dos átomos de hidrógeno conlleva a la formación de dos orbitales moleculares: un orbital molecular enlazante y un orbital molecular antienlazante. En el orden de llenado los orbitales marcados con asterisco y marcados con rojo son los orbitales antienlazantes.
👉 Un orbital molecular enlazante se caracteriza por poseer un nivel energético inferior y en consecuencia una mayor estabilidad que los orbitales atómicos por separado.
👉 Un orbital molecular antienlazante tiene una energía mucho mayor que las de los orbitales atómicos por separado haciéndolo notablemente inestable.
Como el nombre enlazante y antienlazante sugiere, la posición de los electrones en un orbital enlazante conlleva a un enlace covalente estable, mientras que los electrones en un orbital antienlazante conllevan a un enlace inestable. De hecho, la forma de la onda de los dos enlaces es diferente y recuerda a las interferencias constructivas y de interferencia.
Un orbital enlazante es como una onda constructiva, haciendo que la densidad electrónica entre los dos núcleos se refuerce, haciendo que la carga electrostática que une a los núcleos sea más grande formando un fuerte enlace estable. Por el contrario, la densidad electrónica del orbital antienlazante esa destructiva o de interferencia, provocando que la densidad de electrones entre los núcleos sea nula, esto provoca una repulsión por la carga positiva de los núcleos y una atracción cercana a cero, lo que provoca una tendencia a la separación.
21.5 Configuraciones electrónicas en orbitales moleculares
Para entender las propiedades de las moléculas, primero debemos entender cómo es que los electrones se distribuyen en un orbital molecular. El procedimiento para determinar la configuración electrónica de la molécula es análogo al que empleamos para determinar las configuraciones electrónicas de los átomos:
👉 Organizar los orbitales moleculares en orden de energía creciente.
👉 El número de orbitales moleculares es siempre igual al número de orbitales atómicos combinados.
👉 Los orbitales más estables son enlazantes y los más inestables son antienlazantes.
Los orbitales enlazantes se llenan primero al ser más estables.
👉 Al igual que los orbitales atómicos, cada orbital molecular solo puede acomodar dos electrones con spines opuestos en el estado basal de acuerdo al principio de Pauli y de Hund.
👉 El número de electrones en los orbitales moleculares es siempre igual a la suma de todos los electrones de los orbitales atómicos.
21.6 Orden de enlace
El orden de enlace indica la fuerza o estabilidad aproximada de un enlace. El orden de enlace se calcula mediante la siguiente fórmula:

Fórmula del orden de enlace, la cual se lee como: orden de enlace (Od) es igual a un medio (1/2) como factor común de la diferencia entre el número de electrones enlazantes (Neenl) menos el número de electrones antienlazante (Neant).
El orden de enlace indica la fuerza aproximada de un enlace covalente. Por ejemplo, si hay dos electrones en un orbital enlazante y ninguno en el antienlazante, el orden de enlace es uno, lo cual indica que dicho enlace es muy estable. Si el orden de enlace arroja valores de cero o negativos implica que el enlace es inestable o que es repulsivo. El concepto de orden de enlace solo puede usarse cualitativamente para propósitos de determinar la estabilidad de un solo enlace, pero no es útil para comparaciones debido a que el orden de enlace no implica fuerza de enlace.
Debido a la simetría de orbitales enlazantes y antienlazantes, solo debemos calcular el orden de enlace para el último nivel de energía, pues si los niveles inferiores están llenos, cancelaran sus órdenes de enlace.
21.8 Hidrógeno molecular y catión hidrógeno
Vamos a predecir la configuración electrónica por la teoría del orbital molecular y a determinar el orden de enlace. Primero para el gas de hidrógeno.
Ejemplo.
Calcule el orden de enlace y distribuya los electrones de la molécula de dihidrógeno según la teoría de orbital molecular indicando si la molécula es teóricamente estable o inestable.
Cada hidrógeno por separado tiene 1 electrón, por lo que hay 2 electrones para repartir en el siguiente orden de llenado:
σ1s()<σ*1s()<σ2s()<σ*2s()<π2py()=π2pz()<π2px()<π*2py()=π*2pz()<π*2px()
Los repartimos
σ1s(2)
Solo hay electrones para llenar el primer orbital enlazante, ahora calculamos el orden de enlace:

Por lo tanto, podemos concluir que la molécula de dihidrógeno es estable.
Ejemplo.
Calcule el orden de enlace y distribuya los electrones del ion de dihidrógeno(1+) según la teoría de orbital molecular indicando si la molécula es teóricamente estable o inestable.
Cada hidrógeno por separado tiene 1 electrón, pero debido a que es un ión de (1+) asumimos un electrón menos, por lo que hay 1 electrón para repartir en el siguiente orden de llenado:
σ1s()<σ*1s()<σ2s()<σ*2s()<π2py()=π2pz()<π2px()<π*2py()=π*2pz()<π*2px()
Los repartimos
σ1s(1)
Solo hay electrones para llenar el primer orbital enlazante, ahora calculamos el orden de enlace:

El orden de enlace es de 0,5 menos que para el gas de hidrógeno, pero sigue siendo estable.
21.9 Molécula de dihelio
Ejemplo.
Calcule el orden de enlace y distribuya los electrones de la molécula de dihelio según la teoría de orbital molecular indicando si la molécula es teóricamente estable o inestable.
Cada helio por separado tiene 2 electrón, por lo que hay 4 electrones para repartir en el siguiente orden de llenado:
σ1s()<σ*1s()<σ2s()<σ*2s()<π2py()=π2pz()<π2px()<π*2py()=π*2pz()<π*2px()
Los repartimos
σ1s(2) <σ*1s(2)
El orden de enlace:
La teoría del orbital molecular sirve entre otras para comprender porque el helio es noble, es decir estable como un átomo individual e inestable al enlazarse consigo mismo.
La repulsión generada por el orbital antienlazante σ*1s(2) lleno contrarresta la atracción del orbital enlazante σ1s(2), lo cual deja una fuerza de enlace neta de cero, por lo que los átomos de helio no se unirán espontáneamente para formar la molécula de dihelio.
A pesar de ello pueden crearse condiciones para crear el dihelio a las malas, cosa que se logró en 1993, aunque aún bajo condiciones experimentales el dihelio solo tiene una vida muy corta antes de descomponerse.
21.10 Moléculas adiatómicas del segundo periodo.
Aparentemente crear o predecir moléculas mediante la teoría del orbital molecular puede convertirse en un dolor en el trasero, por lo que consideraremos únicamente moléculas muy simples, compuestas por el mismo elemento y solo con dos átomos.
👉 Dicarbono
- Para la molécula de di-carbono (C2) tendríamos 12 electrones a repartir, se aconseja usar un patrón de color para los anti-enlazantes, para evitar confusiones molestas: σ1s(2)σ*1s(2)σ2s(2)σ*2s(2)π2py(2)π2pz(2). Esto genera un orden de enlace de (Od = 0,5 (8 - 4) = 2). Como el orden de enlace es mayor que 1 se espera que el dicarbono sea una sustancia estable, de hecho, esta sustancia ha sido detectada en estado gaseoso con propiedades diamagnéticas al tener todos sus orbitales moleculares llenos. Tenga en cuenta que los dobles enlaces en C2 son ambos enlaces pi debido a los cuatro electrones en los dos orbitales moleculares π. En la mayoría de las otras moléculas, un doble enlace está formado por un enlace sigma y un enlace pi.
👉 Dioxígeno
- Para la molécula de dioxígeno (O2) tendríamos 16 electrones a repartir σ1s(2)σ*1s(2)σ2s(2)σ*2s(2)π2py(2)π2pz(2)π2px(2)π*2py(1) π*2pz(1). Esto genera un orden de enlace de (Od = 0,5 (6 - 2) = 2), se ignoran los orbitales moleculares σ debido a que, al estar llenos completamente, su efecto neto en la estabilidad de la molécula es cero. Dado que se generan dos orbitales anti-enlazantes semi-llenos, se espera que el dioxígeno sea diamagnético.
22. Efecto del enlace químico en los estados de la materia
|| REGRESAR AL INDICE ||
22.1 Enlaces en el estado sólido
La estructura de los sólidos puede distinguirse en dos categorías básicas, la estructura amorfa y la estructura cristalina. El hielo es un sólido cristalino, que posee una estructura rígida y un orden de amplio rango; sus átomos, moléculas o iones ocupan posiciones específicas y rígidas. La organización de los átomos, moléculas o iones en un cristal es tal que la energía atractiva neta de las fuerzas intermoleculares se encuentra a su máximo. Las fuerzas responsables por el mantenimiento de un cristal pueden ser iónicas, covalentes, van des Waals o momentos dipolares (Puentes de hidrógeno); así como una combinación de todas.

Figura 22.1. En los sólidos amorfos como el vidrio, se carece de una estructura bien definida.

Figura 22.2. La sal común es un cristal iónico.
👉 Sólidos amorfos
De cierta forma los sólidos amorfos se comportan de manera semejante a los líquidos, solo que todo más lento. En ese sentido hay estructuras minicristalinas que funcionan como unidades, pero a diferencia de un cristal normal, las unidades no se unen entre sí a largas distancias, lo cual da cierta movilidad a la estructura. Sin embargo, los sólidos amorfos poseen una unión a distancias intermedias, lo cual hace que se muevan muy lento, de lo contrario serían líquidos. De hecho, las uniones a mediana distancia son lo bastante fuertes para mantener el conjunto estático de forma que se impide el movimiento, pero no lo bastante fuertes como para manifestar una estructura ordenada. Un ejemplo de un sólido amorfo es el vidrio, el cual no se mueve, no fluye, pero no posee una estructura organizada. Un elemento puede formar tanto una forma cristalina como una forma amorfa, por lo general muchos sólidos amorfos se generan por un congelamiento rápido del estado líquido, lo cual impide que los átomos tengan tiempo de organizarse en la altamente organizada estructura cristalina, creando el sólido amorfo con estructura líquida pero completamente inmóvil.
👉 Sólidos cristalinos
La estructura y propiedades de los sólidos cristalinos como el punto de fusión, la densidad y la dureza son determinadas por las fuerzas atractivas fuertes y débiles juntas. De hecho, podemos clasificar a los sólidos cristalinos de acuerdo a la fuerza predominante que mantiene unida la estructura.
Cristales iónicos
Los cristales iónicos consisten en iones que se mantienen unidos mediante la fuerza electrostática de los iones. En este sentido el enlace iónico no solo une dos polos electrostáticos bien definidos, sino todos los que le quepan en las tres dimensiones. La estructura de un cristal iónico dependerá de las cargas del catión, del anión y de sus radios. El tamaño del ion afecta el modo entre las otras moléculas iónicas se asocia en las tres dimensiones creando 7 posibles unidades cristalinas altamente organizadas. Los sólidos iónicos se encuentran unidos fuertemente, por lo que poseen puntos de fusión altos, además no conducen la electricidad debido a que los iones y sus electrones se encuentran estructuralmente empaquetados en las unidades cristalinas. Sin embargo, cuando son disueltos en agua o se funden los iones se liberan, lo cual permite un flujo de electrones extremadamente eficiente.
Cristales moleculares
Los cristales moleculares consisten en átomos o moléculas que se mantienen unidos mediante las interacciones moleculares, ya sean por las interacciones entre dipolos o mediante las fuerzas de van der Waals. Un ejemplo de un cristal molecular es el dióxido de azufre sólido, en donde la fuerza atractiva predominante es la fuerza dipolo-dipolo.

Figura 22.3. El hielo es un cristal molecular.
En el caso del hielo, la fuerza atractiva predominante es el puente de hidrógeno, que a su vez es un caso especial de las interacciones dipolo-dipolo. Otros ejemplos de cristales moleculares son el yodo molecular, el fósforo molecular (P4) y el azúfre molecular (S8). Con excepción del hielo, los cristales moleculares se empaquetan tan cerca como les permite el tamaño de sus moléculas y su organización geométrica.
Debido a que las interacciones moleculares son mucho más débiles que el enlace iónico, los sólidos moleculares poseen puntos de fusión más bajos que los cristales iónicos, y de hecho muchos de ellos se funden a temperaturas menores a los 200°C.
Cristales covalentes
Los cristales covalentes, también denominados como redes cristalinas covalentes, son átomos que se mantienen unidos por enlaces covalentes en una red extensa en tres dimensiones. No puede señalarse la presencia de una molécula única; pero si puede ocurrir que un mismo elemento presente diversas maneras de empaquetarse. Un ejemplo típico es el carbono, que puede unirse a otros carbonos formando diferentes cristales, que en suma forman varios alótropos, los alótropos vistos macroscópicamente parecen materiales diferentes, para el carbono estos materiales aparentemente diferentes son el grafito, el carbón mineral, el diamante y el grafeno.

Figura 22.4. A parte de los cristales naturales grafito y diamante, se ha logrado crear otros como los nanotubos y las esferas de carbono.
En el diamante los carbonos se organizan tetraédricamente mediante enlaces simples a otros cuatro carbonos. Estos enlaces sigma perfectos en tres dimensiones contribuyen a la dureza del diamante (Material natural más duro conocido) y su alto punto de fusión (3550°C). En el grafito los átomos de carbono se organizan en anillos de seis mediante enlaces sigma y enlaces pi. El enlace pi es menos estable que el sigma, y de forma análoga a lo que sucede en el benceno, los electrones pi tienden a deslocalizarse y moverse con facilidad. Para el caso del grafito, los electrones pi pueden moverse fácilmente, haciendo del grafito un buen conductor de la electricidad. Debido a que las unidades del grafito son planas, por lo que aun cuando dichas hojas sean duras, se pueden mover una sobre la otra de forma sencilla, esto le da al grafito sus características únicas como material para escritura además de ser un buen lubricante.
Otro tipo de cristal covalente es el cuarzo creado por moléculas de dióxido de silicio. La organización de los átomos de silicio en el cuarzo es similar a las del carbono en el diamante, pero debido a la presencia de oxígeno, el enlace covalente es imperfecto, por lo que el cuarzo presenta propiedades semejantes, pero no tan extremas como las del diamante.
Cristales metálicos
De cierta forma los cristales metálicos son los más fáciles de estudiar debido a que cada vértice del cristal está ocupado por un átomo simple del mismo elemento. El enlace de los metales es bastante diferente del de otros cristales. En el enlace de los metales no hay enlaces individuales que unan a cada unidad, sino un colosal enlace púnico compuesto por electrones deslocalizados que rodean a la red de átomos metálico. De hecho, los metales pueden imaginarse como una red de iones positivos inmersos en una red o nube de electrones deslocalizados, prestos para ser captados por átomos oxidantes. La gran fuerza cohesiva de esta nueve de electrones es la responsable por la gran fuerza de los metales, que aumenta con el número de electrones de valencia.
Por ejemplo, los metales con un solo electrón de valencia tienen puntos de fusión relativamente bajo (97.6°C) y son blandos, mientras que el aluminio que posee tres electrones de valencia tiene un punto de fusión de 660°C y es más duro. Cabe anotar que esta tendencia no se cumple tan bien para los metales de transición con valencias variables, el hierro por ejemplo tiene valencias 2 y máximo 3, pero posee un punto de fusión de 1558°C. la mayoría de los metales de transición son duros y poseen altos puntos de fusión (Chang & Overby, 2011; Chang, 2006; Ebbing & Gammon, 2008; McMurry et al., 2007; Timberlake, 2015).
La movilidad de los electrones deslocalizados del enlace metálico los hace fáciles de empujar y agitar, lo cual les otorga sus conocidas propiedades conductivas de electricidad y calor, aunque también debido a la deslocalización, los electrones metálicos de valencia son fáciles de capturar por elementos oxidantes, lo cual los hace susceptibles a la oxidación.
22.2 Enlaces en el estado líquido y gaseoso
El estado líquido se caracteriza por un movimiento de las moléculas relativamente libre y relativamente organizado. En términos moleculares es un estado amorfo pero con una capacidad atractiva intermedia y baja. Los líquidos se forman cuando se adiciona suficiente energía a un sólido, lo cual desestabiliza las fuerzas que lo mantiene unido. Mientras más débiles sean dichas fuerzas, menos energía es necesaria para desestabilizar los componentes del sólido.
Una vez en estado líquido las moléculas interactúan entre sí, tanto atractiva como repulsivamente, provocando las propiedades de flujo, incompresibilidad y adquisición de la forma del contenedor donde se encuentren. En el estado gaseoso se adiciona suficiente energía al sistema como para romper todas las interacciones moleculares, lo cual hace que las moléculas se muevan independientes unas de otras, incrementando al máximo el volumen que ocupan, ya que no hay fuerza que las mantengan unidas. Hay que resaltar que aquellas moléculas que no presentan momentos dipolares son más susceptibles a separarse.
Referencias bibliográficas
|| REGRESAR AL INDICE ||
Bader, R. F. W., Hernández‐Trujillo, J., & Cortés‐Guzmán, F. (2007). Chemical bonding: from Lewis to atoms in molecules. Journal of Computational Chemistry, 28(1), 4–14.
Bakowies, D., & Thiel, W. (1996). Hybrid models for combined quantum mechanical and molecular mechanical approaches. The Journal of Physical Chemistry, 100(25), 10580–10594.
Bantz, D. A. (1980). The structure of discovery: Evolution of structural accounts of chemical bonding. In Scientific Discovery: Case Studies (pp. 291–329). Springer.
Bevan, D. J. M., & Hagenmuller, P. (2013). Non-stoichiometric compounds: tungsten bronzes, vanadium bronzes and related compounds. Elsevier.
Branch, G. E. K. (1984). Gilbert Newton Lewis, 1875-1946. ACS Publications.
Brown, T. L., LeMay, H. E. J., Bursten, B. E., Murphy, C. J., & Woodward, P. (2009). Chemistry the central science (11th ed.). Pearson; Prentice Hall.
Brown, T. L., LeMay, H. E. J., Bursten, B. E., Murphy, C. J., Woodward, P. M., & Stoltzfus, M. W. (2017). Chemistry, the central science (13th ed.). Boston: Pearson.
Brown, T. L., LeMay, H. E. J., Bursten, B. E., Murphy, C. J., Woodward, P., & Stoltzfus, M. W. (2015). Chemistry the Central Science.
Chang, R. (2006). Chang’s “General Chemistry - Essential Concepts” (4th ed.). McGraw-Hill New York.
Chang, R. (2010). Chemistry (10th ed.). McGraw-Hill New York.
Chang, R., & Overby, J. (2011). General Chemistry,Th e Essential Concepts (11th ed.). McGraw-Hill New York.
Christe, K. O. (2013). Bartlett’s discovery of noble gas fluorides, a milestone in chemical history. Chemical Communications, 49(41), 4588–4590.
Cremer, D., & Kraka, E. (1984). A description of the chemical bond in terms of local properties of electron density and energy. Croat. Chem. Acta, 57(6), 1259–1281.
Dunitz, J. D., & Gavezzotti, A. (2009). How molecules stick together in organic crystals: weak intermolecular interactions. Chemical Society Reviews, 38(9), 2622–2633.
Ebbing, D. D., & Gammon, S. D. (2008). General chemistry. (Houghton Mifflin Company, Ed.) (9th ed.). Bonston.
Ebbing, D. D., & Gammon, S. D. (2017). General chemistry (11th ed.). Boston: Cengage Learning.
Eliade, M., & Ledesma, M. P. (1974). Herreros y alquimistas. Alianza Madrid.
Harris, H. H. (1999). A Biography of Distinguished Scientist Gilbert Newton Lewis. Journal of Chemical Education, 76(11), 1487.
Hund, F. (1977). Early history of the quantum mechanical treatment of the chemical bond. Angewandte Chemie International Edition in English, 16(2), 87–91.
Jensen, W. B. (1984). Abegg, Lewis, Langmuir, and the octet rule. Journal of Chemical Education, 61(3), 191.
Kohler, R. E. (1971). The origin of GN Lewis’s theory of the shared pair bond. Historical Studies in the Physical Sciences, 3, 343–376.
Kuhn, H. (1945). An Essay on Man. An Introduction to a Philosophy of Human Culture. The Journal of Philosophy, 42(18), 497–504.
Larder, D. F. (1971). A Dialectical Consideration of Butlerov’s Theory of Chemical Structure. Ambix, 18(1), 26–48.
Lennard-Jones, J. (1949). The molecular orbital theory of chemical valency. ii. equivalent orbitals in molecules of known symmetry. In Proceedings of the Royal Society of London A: Mathematical, Physical and Engineering Sciences (Vol. 198, pp. 14–26). The Royal Society.
Levere, T. H. (2001). Transforming matter: a history of chemistry from alchemy to the buckyball. JHU Press.
Lindsay, S. (2009). Introduction to nanoscience. OUP Oxford.
Mackay, A. L. (2002). Generalized crystallography. Structural Chemistry, 13(3–4), 215–220.
McMurry, J., Castellion, M. E., & Ballantine, D. S. (2007). General, Organic and Biological chemestry, 5ed. Pearson; Prentice Hall.
McMurry, J., Fay, C. R., & Fantini, J. (2012). Chemistry (6th ed.). Boston: Pearson.
Niaz, M. (2001). A rational reconstruction of the origin of the covalent bond and its implications for general chemistry textbooks. International Journal of Science Education, 23(6), 623–641.
Pyle, A. (1995). Atomism and its critics: from Democritus to Newton.
Rhodes, C. J., & Macrae, R. M. (2015). “Vibrational bonding”: a new type of chemical bond is discovered. Science Progress, 98(1), 12–33.
Soukup, R. W. (2005). Historical aspects of the chemical bond. chemical relationality versus physical objectivity. Monatshefte Für Chemie/Chemical Monthly, 136(6), 803–818.
Timberlake, K. C. (2015). Chemistry An Introduction to General, Organic, and Biological Chemistry (15th ed.). USA: Pearson.
Timberlake, K. C., & Orgill, M. (2019). General, Organic, and Biological Chemistry (6th ed.). San Francisco: Pearson.
Ejercicios resueltos
|| REGRESAR AL INDICE ||
👉 Demostraciones <Ejercicios analíticos para demostrar las respuestas analíticas o algoritmos de solución> (Pulse aquí)
👉 Ejemplos <Ejercicios Propios de la página> (Pulse aquí)
👉 Matamala y González <Libro de texto Ediciones Cultural - 1976> (Pulse aquí)
👉 Química general de Chang <Libro de texto de Raymond Chang> (Pulse aquí)
👉 Química la ciencia central <Libro de texto de Theodore E. Brown> (Pulse aquí)
Demostraciones
Ejemplos
Ejemplo.Dibujar la estructura de Lewis del NaCl
Ejemplo.Dibujar la estructura de Lewis del CaO
Ejemplo.Dibujar la estructura de Lewis del Ca3N2
Ejemplo.Dibujar la estructura de Lewis para Fe2O3
Ejemplo.Dibujar la estructura de Lewis para FeO
Ejemplo.Dibujar la estructura de Lewis para H2O
Ejemplo.Dibujar la estructura de Lewis para NI3
Ejemplo.Dibujar la estructura de Lewis para CO
Ejemplo.Dibujar la estructura de Lewis para H2O2
Ejemplo.Dibujar la estructura de Lewis para NaClO
Ejemplo.Dibujar la estructura de Lewis sin octeto extendido para H2SO4
Ejemplo.Dibujar la estructura de Lewis con octeto expandido para H2SO4.
Ejemplo.Dibujar la estructura de Lewis sin octeto extendido para H3PO4
Ejemplo.Dibujar la estructura de Lewis con octeto expandido para H3PO4.
Ejemplo.Determinar las posibles estructuras de Lewis y su representación de resonancia.
Ejemplo.Determinar las estructuras de resonancia el anión NO3(-)
Ejemplo.Dibujar la estructura de Lewis para sin octeto extendido Na2SO4
Ejemplo.Dibujar la estructura de Lewis con octeto expandido para Na2SO4.
Ejemplo.Dibujar la estructura de Lewis para PF5
Ejemplo.Dibujar la estructura de Lewis del CO2
Ejemplo.Dibujar la estructura de Lewis del Cl2O
Ejemplo.Dibujar la estructura de Lewis del Cl2O3
Ejemplo.Dibujar la estructura de Lewis del Cl2O5
Ejemplo.Dibujar la estructura de Lewis del Cl2O7
Ejemplo.Dibujar la estructura de Lewis del SO
Ejemplo.Dibujar la estructura de Lewis del SO2
Ejemplo.Dibujar la estructura de Lewis del SO6
Ejemplo.Dibujar la estructura de Lewis del H2CO3
Ejemplo.Dibujar la estructura de Lewis del H2SO2
Ejemplo.Dibujar la estructura de Lewis del H2SO3 sin octeto expandido
Ejemplo.Dibujar la estructura de Lewis del H2SO3 con octeto expandido
LibreChem
Química General de Chang
Química la Ciencia Central
Muestra 8.1. Ordene los compuestos iónicos NaF, CsI y CaO en orden creciente de energía reticular.
Brown13 Práctica 8.1.1. Determine cual de los siguientes órdenes de energía reticular es correcto. (a) NaCl > MgO > CsI > ScN, (b) ScN > MgO > NaCl > CsI, (c) NaCl > CsI > ScN > CaO, (d) MgO > NaCl > ScN > CsI, (e) ScN > CsI > NaCl > MgO.
Práctica 8.1.2. ¿Qué sustancia espera que tenga la mayor energía reticular: MgF2, CaF2 o ZrO2?
Muestra 8.2. Prediga el ion generalmente formado por (a) Sr, (b) S, (c) Al
Muestra 8.2.1. ¿Cuál de estos elementos es más probable que forme iones con una carga de 2+? (a) Li, (b) Ca, (c) O, (d) P, (e) Cl.
Muestra 8.2.2. Prediga las cargas de los iones formados cuando el magnesio reacciona con el nitrógeno.
Muestra 8.4a. ¿Cuál enlace es más polar? B - Cl o C - Cl. Indique en cada caso qué átomo tiene la carga negativa parcial.
Muestra 8.4b. ¿Cuál enlace es más polar? P - F o P - Cl. Indique en cada caso qué átomo tiene la carga negativa parcial.
Práctica 8.4.1. ¿Cuál de los siguientes enlaces es el más polar? (a) H-F, (b) H-I, (c) Se-F, (d) N-P, (e) Ga-Cl.
Práctica 8.4.2. ¿Cuál de los siguientes enlaces es más polar: S-Cl, S-Br, Se-Cl o Se-Br?
Muestra 8.6. Dibuje la estructura de Lewis para el tricloruro de fósforo, PCl3.
Práctica 8.6.2. (a) ¿Cuántos electrones de valencia deberían aparecer en la estructura de Lewis para CH2Cl2? (b) Dibuje la estructura de Lewis.
Muestra 8.7. Dibujar la estructura de Lewis para HCN
Práctica 8.7.2. Dibuje la estructura de Lewis para (a) ion NO+, (b) C2H4.
Muestra 8.8. Dibuje la estructura de Lewis para el ion BrO3-.
Práctica 8.8.1. ¿Cuántos pares de electrones no enlazantes hay en la estructura de Lewis del ion peróxido, O22-? (a) 7, (b) 6, (c) 5, (d) 4, (e) 3.
Práctica 8.8.2. Dibuje la estructura de Lewis para (a) ClO2-, (b) PO43- .
Ejemplo página 318. Determinar la estructura de Lewis dominante para el CO2 (dadas en la figura).
Muestra 8.9. Tres posibles estructuras de Lewis para el ion tiocianato, NCS-, estan dadas en la figura adjunta. Determinar cual es la estructura de Lewis dominante. (pulse en la figura)
Muestra 8.10. ¿Cuál se prevé que tenga los enlaces azufre-oxígeno más cortos, SO3 o SO32- ?
Práctica 8.10.2. Dibuje dos estructuras de resonancia equivalentes para el ion formiato, HCO2-.
No hay comentarios:
Publicar un comentario